Whole Genome Sequencing (WGS) has revolutionized bacterial strain typing in molecular epidemiology. In silico MLST, SNP analysis, and gene-by-gene analysis have emerged as key methods. This article explores how WGS-based approaches, including core genome MLST (cgMLST), are replacing traditional molecular typing techniques like PFGE and MLVA in public health and food safety.
Whole Genome Sequencing (WGS)-Based Analysis: Revolutionizing Molecular Epidemiology
in silico MLST: A Breakthrough in Bacterial Strain Typing
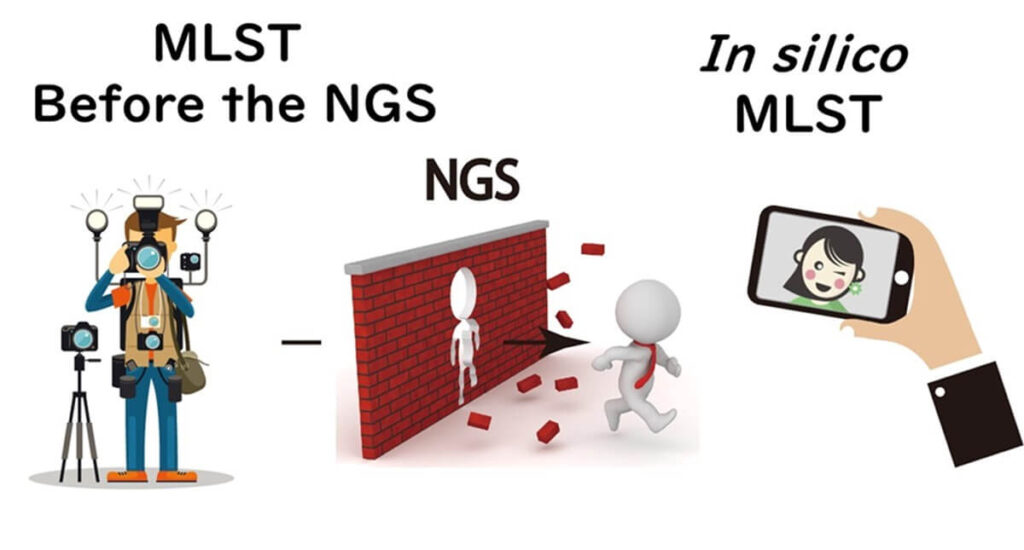
The advent of next-generation sequencing (NGS) has transformed Multi-Locus Sequence Typing (MLST) by shifting from conventional PCR-based sequencing to in silico MLST. Traditionally, MLST required individual amplification and sequencing of seven genes per strain. Now, with NGS, whole genome sequences can be analyzed computationally, eliminating the need for labor-intensive PCR.
Note: "In silico" refers to computer-based analysis, similar to "in vivo" (within a living organism) and "in vitro" (in a test tube or glassware).
Since its introduction in 2000, MLST has been widely used in bacterial molecular epidemiology. Over the past two decades, researchers—including those in my lab—have extracted and sequenced genes manually. However, NGS has streamlined this process, allowing bacterial isolates to be sequenced in bulk and analyzed using specialized online databases that automatically determine in silico MLST types. This advancement significantly reduces processing time and labor.
Public Health Applications of in silico MLST
Public Health England (PHE) adopted in silico MLST for Salmonella serotyping in 2015, utilizing Whole Genome Sequencing (WGS). The approach leverages MLST with seven genes to determine serotypes, making WGS-based typing practical for routine public health applications.
For a more detailed explanation, refer to:
Determining Salmonella Serotypes with Next-Generation Sequencers: In Silico MLST in the UK since 2015
in silico MLST for Food Safety: A Practical Guide
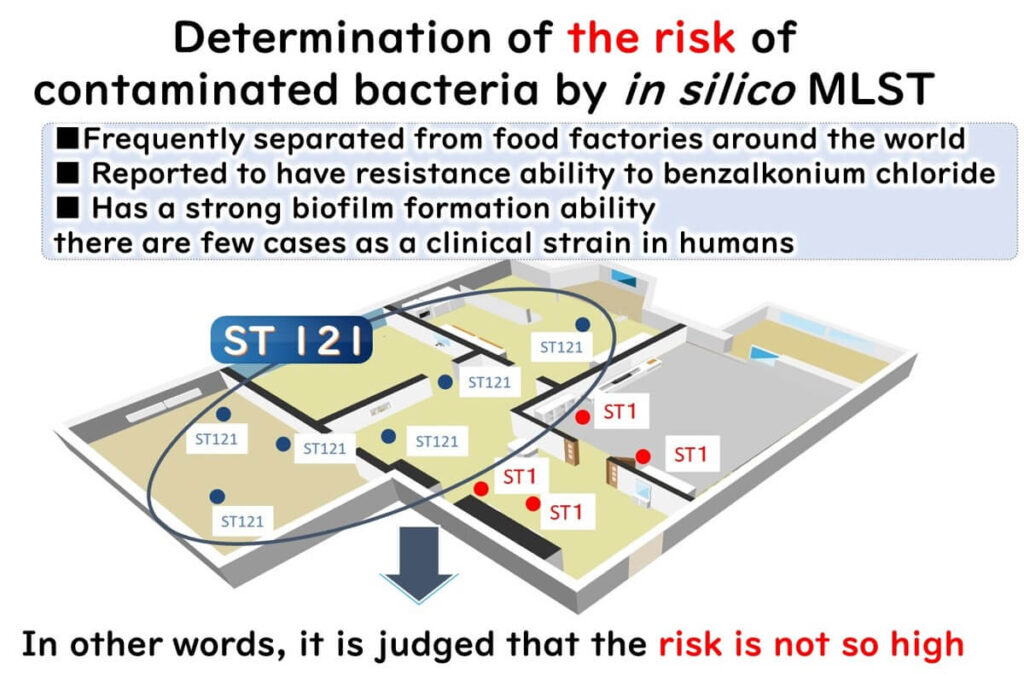
In food factories, Listeria monocytogenes contamination is a significant concern. By applying in silico MLST, bacterial isolates can be classified into Sequence Types (STs), which provide critical epidemiological insights.
- Example:
- ST221 is commonly detected in food factories globally but is not highly virulent.
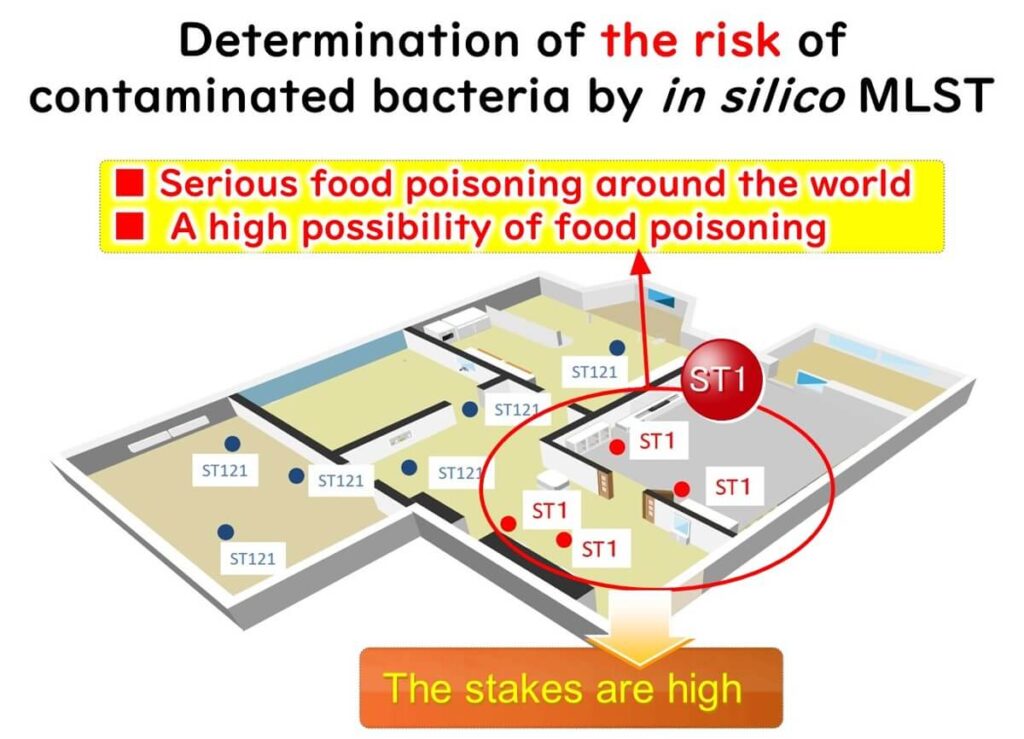
- ST1, however, is associated with severe listeriosis outbreaks and high mortality rates worldwide.
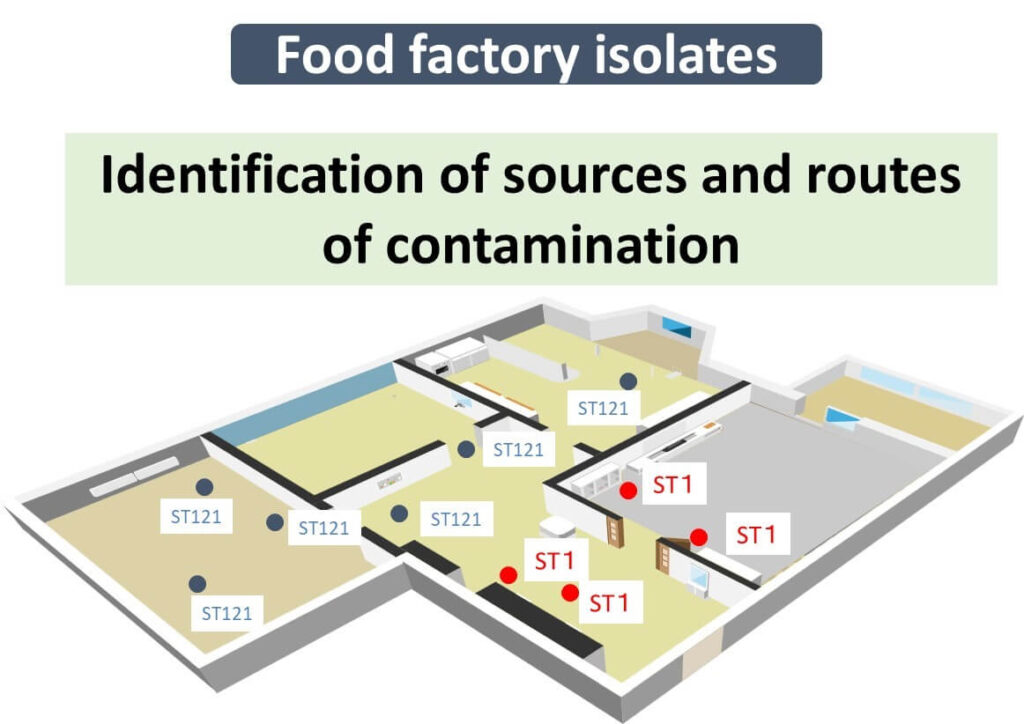
Identifying ST1 in a facility requires immediate sanitation measures, including deep cleaning and sterilization, to prevent potential foodborne illness outbreaks.
📌 Key Insight:
In silico MLST enables food manufacturers to implement data-driven risk management strategies, ensuring food safety and compliance with global regulatory standards.
The Future of in in silico MLST in Bacterial Typing
With the rise of core genome MLST (cgMLST), the role of in silico MLST is evolving. Future trends will focus on:
- Scalability: Increased global adoption for foodborne pathogen surveillance.
- Automation: Integration with AI-driven sequencing platforms.
- Regulatory Compliance: Standardization across public health agencies worldwide.
For an in-depth analysis, see:
Will the emergence of core genome MLST end the role of in silico MLST?
Whole Genome Sequencing (WGS) for Molecular Epidemiology
Aside from in silico MLST, the most widely used molecular epidemiology techniques involving WGS can be broadly categorized into two main approaches:
- Single Nucleotide Polymorphism (SNP) Analysis
- Gene-by-Gene An

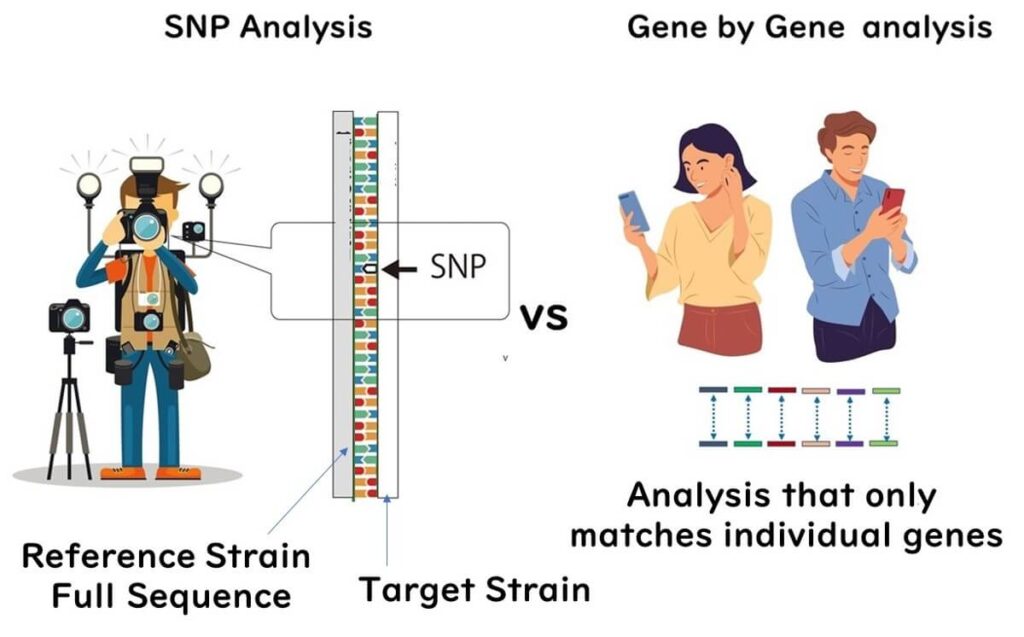
Understanding SNP Analysis in WGS
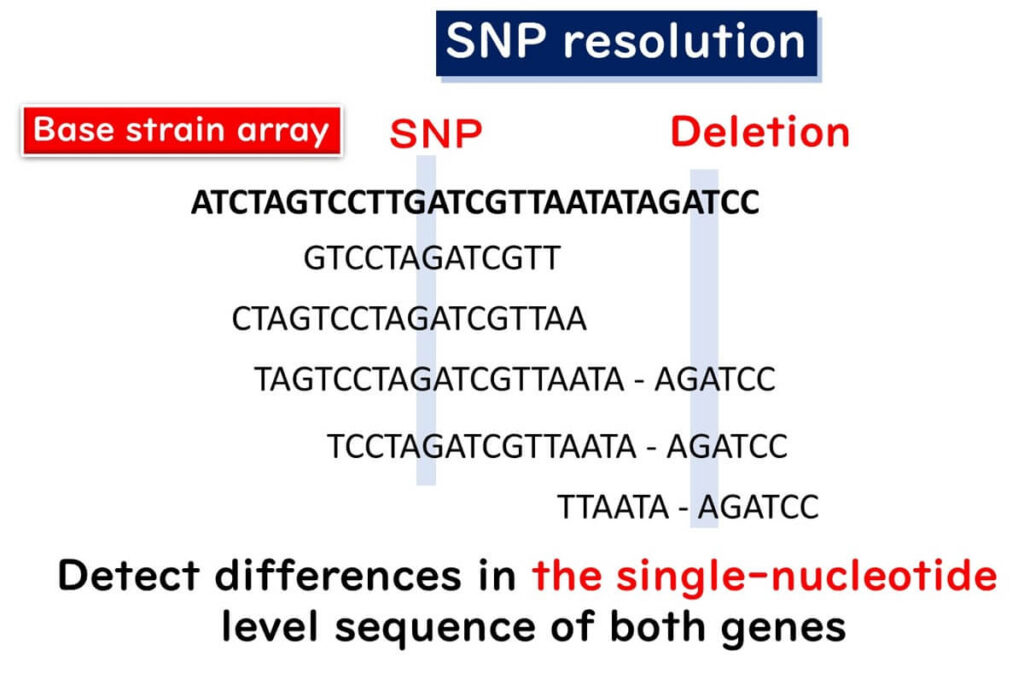
SNP analysis in whole genome sequencing (WGS) identifies strain differences based on single nucleotide polymorphisms (SNPs) across the entire genome. This method requires aligning the genome sequence of the sample with a reference genome, making it a powerful tool for molecular epidemiology.
SNPs serve as highly informative genetic markers, helping to elucidate the evolutionary history of homogeneous bacterial groups. Over the past few years, SNP-based molecular epidemiology has been widely used for tracking Salmonella foodborne outbreaks in many countries.
In SNP analysis, any single nucleotide difference between the sample and the reference strain is identified as an SNP mutation. This includes nucleotide deletions detected in the reference genome.
Key Steps in SNP-Based Typing
- Alignment of short reads obtained from next-generation sequencing (NGS) to a reference genome.
- Identification of SNPs that differentiate the sample from the reference strain.
- Construction of phylogenetic trees based on SNP variation to determine the relatedness of bacterial strains.
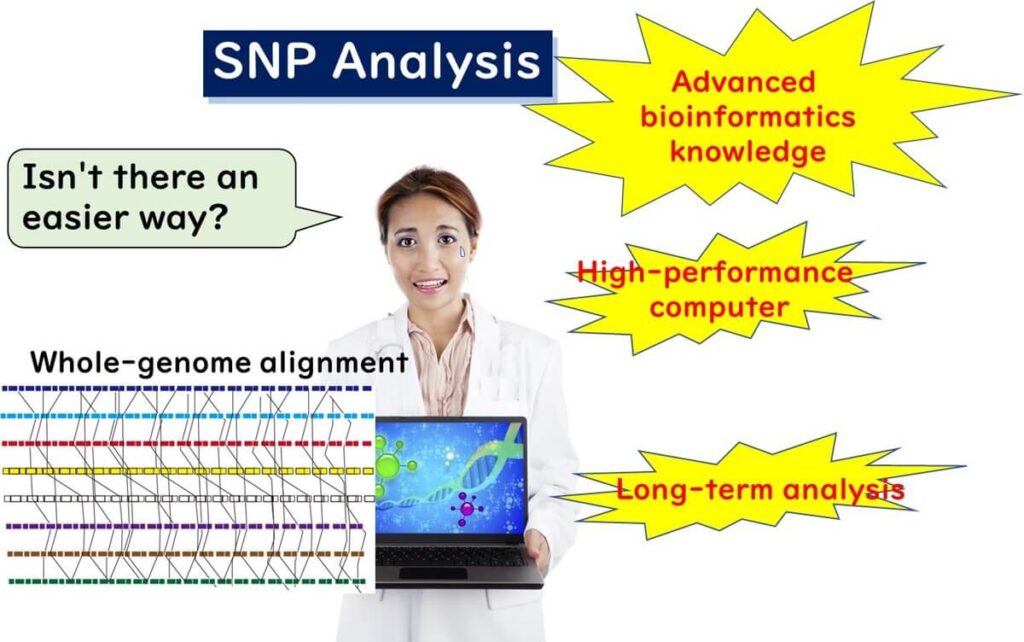
What is Alignment in WGS?
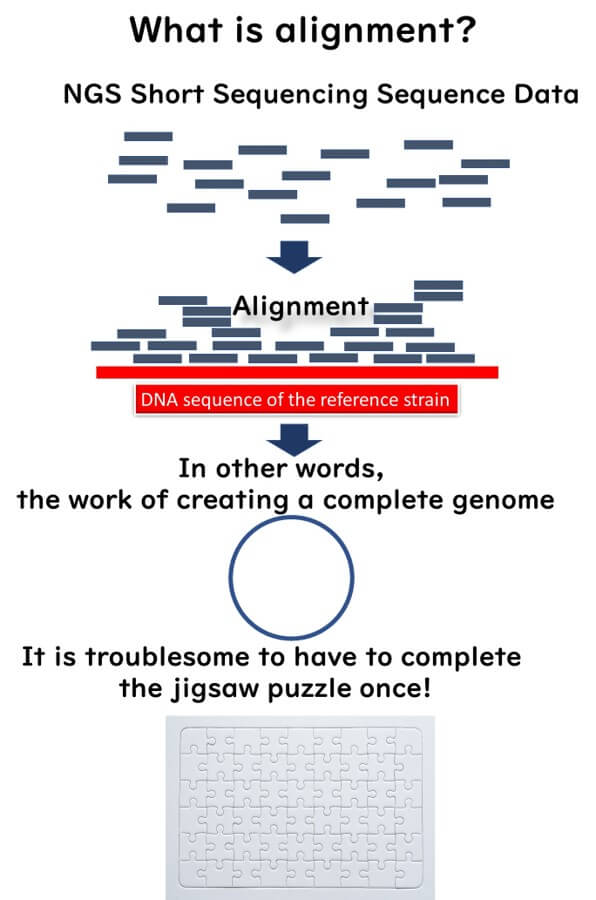
SNP-based molecular epidemiology relies heavily on alignment. The short DNA fragments sequenced by NGS are mapped to a reference genome, a process known as genome alignment. This allows for precise comparison of nucleotide variations across bacterial strains.
SNP analysis is akin to solving a jigsaw puzzle, where the short sequencing reads are assembled into a complete genome before identifying SNPs. However, this process is:
- Labor-intensive
- Time-consuming
- Requires bioinformatics expertise
SNP Analysis: Pros and Cons for Bacterial Typing
Limitations of SNP Analysis
- Complexity – Requires alignment with a reference genome, demanding high-performance computing and advanced bioinformatics expertise.
- Limited data sharing – SNP-based typing is challenging to standardize across laboratories.
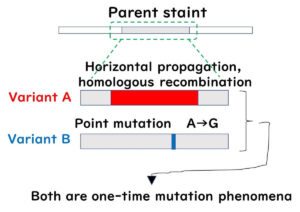
- Overestimation of genetic distances – Horizontal gene transfer and homologous recombination may distort phylogenetic analysis.
Advantages of SNP-Based Typing
✅ High-resolution typing – SNP analysis provides fine-scale differentiation of bacterial strains.
✅ Useful for outbreak investigations – Helps identify closely related bacterial isolates.

Overcoming the Challenges of SNP Analysis
To facilitate NGS-based molecular epidemiology, there is a growing need for:
- More user-friendly bioinformatics pipelines
- Web-based SNP analysis tools that simplify genome alignment
- Standardized SNP databases for global comparison
Gene-by-Gene Analysis: A Practical Alternative to SNP-Based Typing
In 2013, a groundbreaking study introduced gene-by-gene analysis as a novel approach in bacterial molecular epidemiology using next-generation sequencing (NGS). Developed by Professor Martin Maiden of Oxford University, the pioneer behind MLST, this method allows for analyzing short sequence fragments obtained from NGS without the need for alignment to a reference strain. Instead, it directly compares genes within the microbial genome on a computer, essentially performing MLST analysis on a gene-by-gene basis. This technique has since gained widespread adoption in epidemiological studies, particularly for its ease of implementation and data comparability across laboratories.
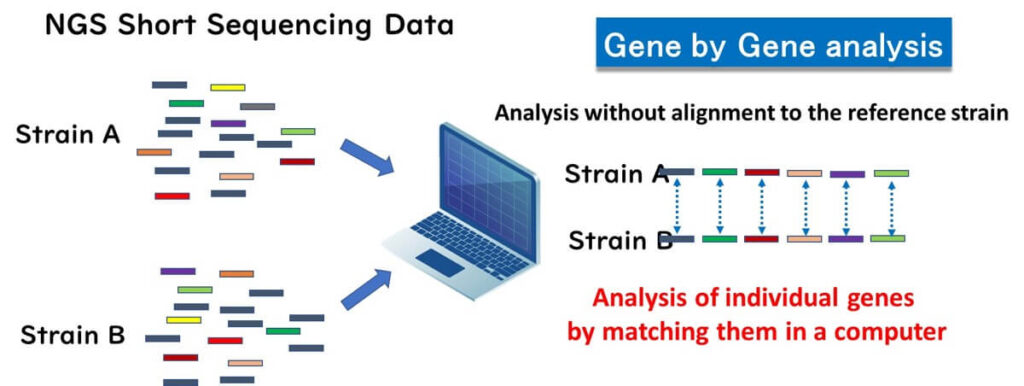
Gene-by-Gene Analysis Method: How It Works
✅ De Novo Assembly: Short-read fragments generated from NGS are assembled into contigs (continuous sequences) using computational tools, eliminating the need for reference genome alignment. This approach significantly reduces bioinformatics complexity and accelerates data processing.
✅ Gene-by-Gene Comparison: Individual genes within the bacterial genome are directly compared across strains on a computer, providing a scalable method for molecular epidemiology and facilitating inter-laboratory data standardization.

Why Gene-by-Gene Analysis is More Practical
✅ 1. No Reference Genome Required: Unlike SNP analysis, gene-by-gene analysis eliminates the need for alignment to a reference genome, making it more accessible to researchers without advanced bioinformatics expertise.
✅ 2. Enhanced Accuracy with Minimal Interference: This method minimizes the impact of homologous recombination and horizontal gene transfer, leading to more precise epidemiological assessments.
✅ 3. Optimized Data Sharing and Reproducibility: Gene-by-gene analysis facilitates seamless data exchange across research institutions, ensuring consistency in molecular epidemiology.
✅ 4. Versatility Across Outbreak Investigations: This approach enables retrospective and real-time outbreak investigations, allowing researchers to compare strains from different timeframes.
Whole Genome Sequencing (WGS)-Based Bacterial Typing
Whole Genome MLST (wgMLST): Expanding Beyond Traditional MLST
Professor Martin Maiden, who introduced MLST, later proposed whole genome MLST (wgMLST) as a gene-by-gene analysis approach. With the advent of next-generation sequencing (NGS), limiting MLST to just seven genes became unnecessary. Thus, wgMLST analyzes all genes in a microorganism’s entire genome.
A typical microorganism has approximately 2,000 to 3,000 genes. Utilizing sequence data from all these genes in MLST provides much higher resolution than the traditional seven-gene method, making it particularly useful for distinguishing between closely related strains.
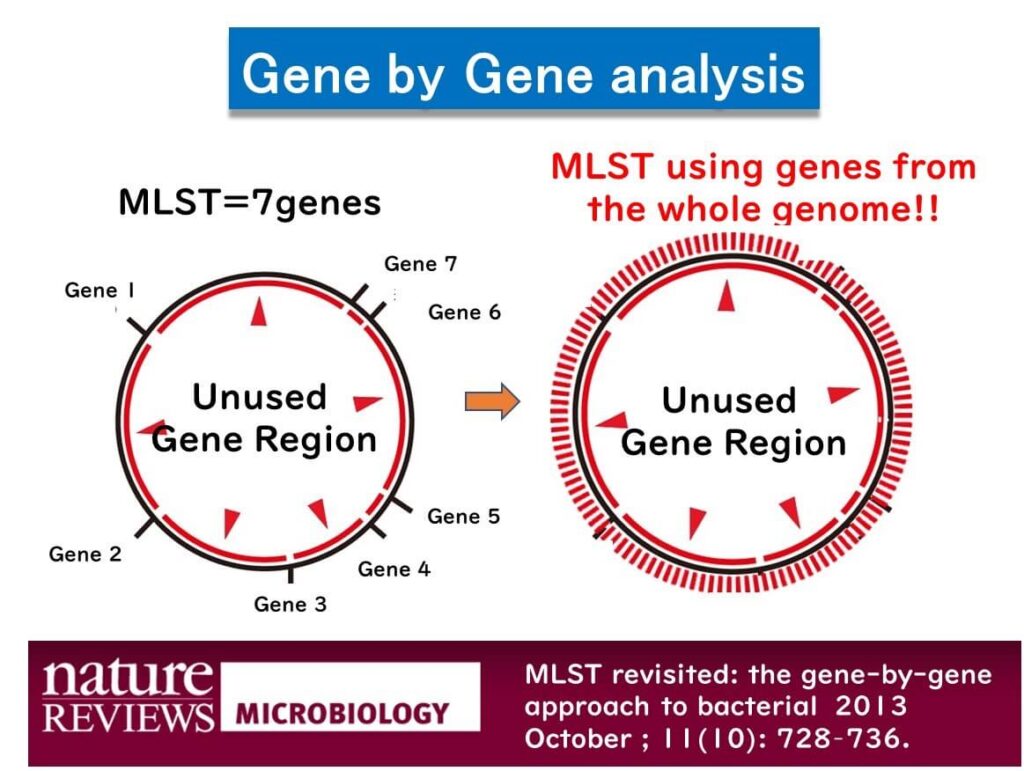
However, wgMLST also has some limitations. Since it targets all the genes in a bacterial strain’s genome, some strains may lack specific genes, which are referred to as the accessory genome. While wgMLST is highly effective for identifying very closely related clones, it becomes problematic when comparing genetically distant strains, as the absence of certain genes makes evaluation inconsistent.
To address these challenges, core genome MLST (cgMLST) was developed.
Core Genome MLST (cgMLST): A Scalable Approach
Core genome MLST (cgMLST) focuses only on the core genes shared by all strains of a species, typically 1,500 to 2,500 genes. By limiting the analysis to these core genes, cgMLST enhances universality and scalability compared to wgMLST, which includes the accessory genome.
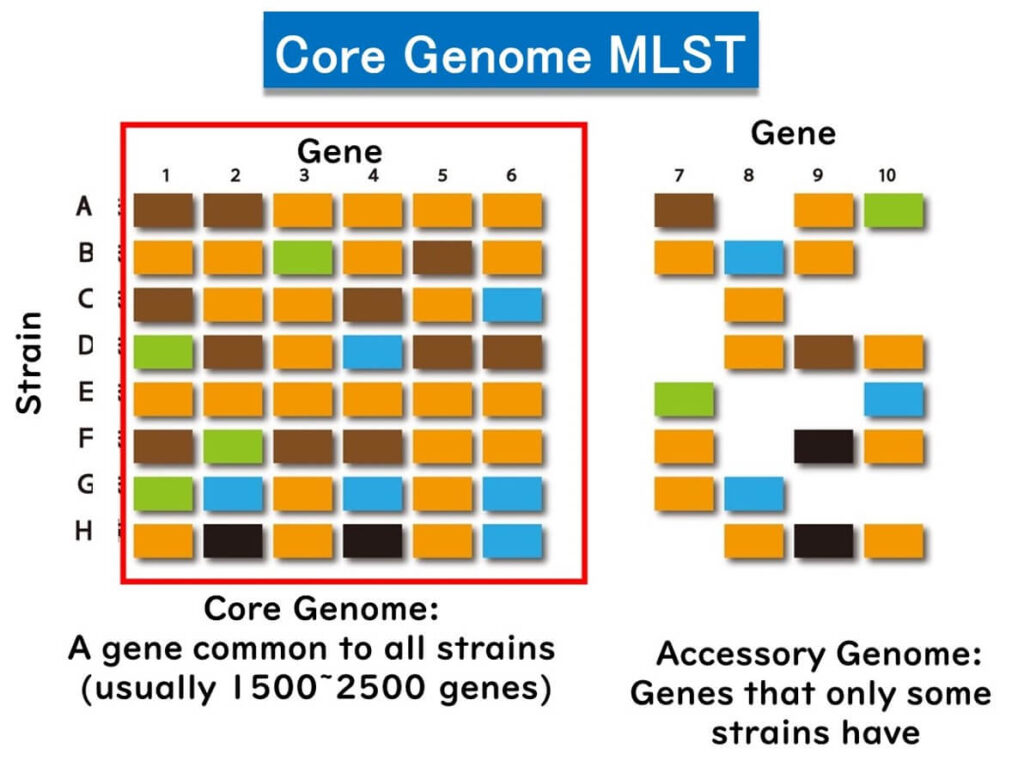
Benefits of cgMLST for Molecular Epidemiology
- No Need for Alignment – Unlike SNP analysis, cgMLST does not require alignment with a reference genome, reducing the need for advanced bioinformatics knowledge.
- Reduced Impact of Homologous Recombination and Horizontal Gene Transfer – Focusing only on core genes minimizes the effects of these phenomena.
- Better Data Sharing – Results are easier to share and compare across research institutions.
- Versatility – cgMLST enables the re-analysis of data for different outbreaks, facilitating the investigation of both new and ongoing cases.
In 2015, cgMLST was introduced based on the idea that analyzing only core genes would provide more consistent and scalable results.
In 2018, Dr. Maiden and his research team at Oxford University applied cgMLST to analyze a large-scale outbreak of Salmonella Enteritidis in Europe. They found that cgMLST had sufficient resolution to track outbreaks across multiple countries and provided reproducible typing results. The outcomes of cgMLST were consistent with SNP analysis, but cgMLST had the added benefit of being easier to standardize across different laboratories due to its reliance on conserved core genes. This method demonstrated high reproducibility and maintained backward compatibility with earlier analyses conducted within the same laboratory.
Furthermore, the emergence of web-based analysis platforms has reduced the need for complex bioinformatics installations, making advanced genome analysis more accessible.
Given these advantages, cgMLST is expected to become the dominant molecular epidemiological tool in the coming decades.
Future Outlook
The rapid advancement of NGS technology is comparable in impact to the emergence of PCR technology 25 years ago. Agencies like the US FDA and CDC have already adopted WGS for foodborne pathogen analysis, replacing older typing techniques like MLST, MLVA, and PFGE. This trend is expected to continue globally.
Currently, the two leading molecular epidemiological methods for tracking foodborne illnesses are:
- SNP Analysis
- cgMLST/wgMLST
Both methods offer high precision and are likely to remain essential tools for public health agencies.

The Future of WGS in Foodborne Pathogen Surveillance
In silico MLST and cgMLST are particularly well-suited for food factories, as they allow:
- Routine monitoring of contamination patterns.
- Easy integration into online genomic databases for data sharing.
- Scalability and accessibility, similar to how 16S rDNA sequencing revolutionized microbiology in the 2000s.
Given these advantages, cgMLST is expected to become the dominant molecular epidemiological tool in the coming decades.