Introduction to Food Microbiology and Safety
Streamlined Molecular Methods for Microbial Identification: A Practical Guide
Molecular methods have revolutionized microbial identification, offering precise tools to define bacterial species and uncover operational taxonomic units (OTUs). In this guide, we demystify the complexities of techniques like 16S rRNA and 16S rDNA sequencing, explaining their significance in identifying species and their role in phylogenetic analysis. From understanding housekeeping genes to selecting ideal genetic regions for analysis, we’ll also delve into the benefits, limitations, and practical considerations. Join us as we explore these powerful tools for microbiological testing, tailored for professionals in the food and safety industries.
The Purpose of Molecular Methods in Food Microbial Testing
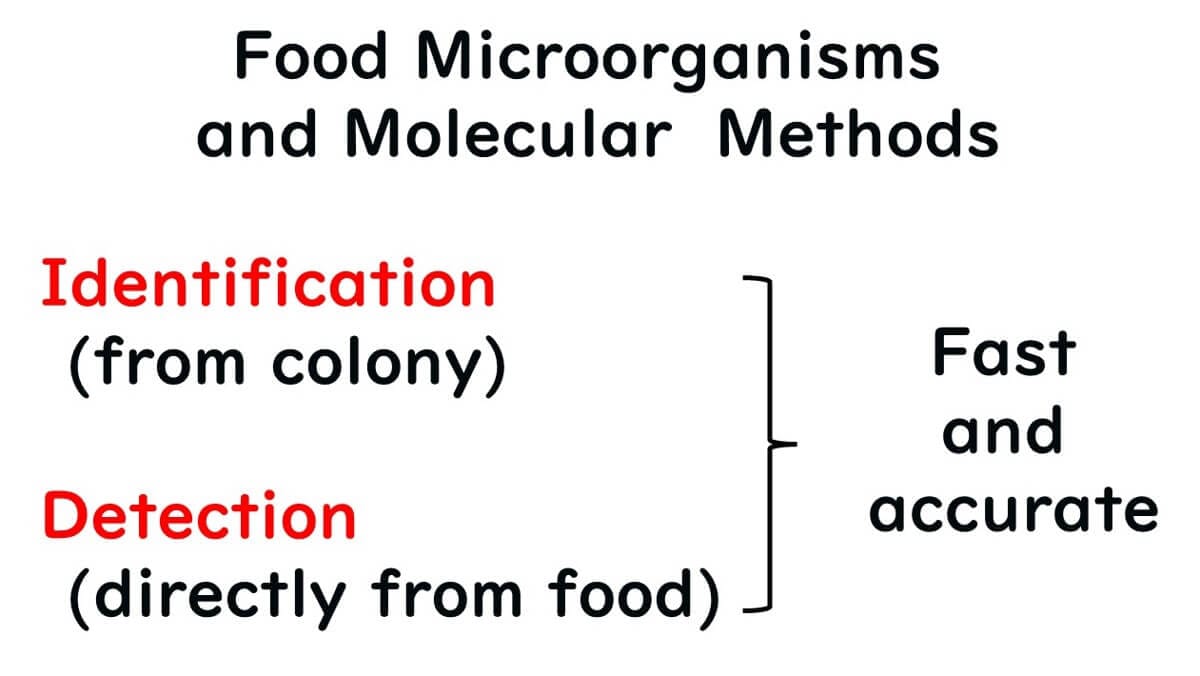
Molecular methods play two pivotal roles in food microbial testing:
1. Identification of isolated bacterial species.
2. Direct detection of specific target bacteria from food samples.
This article focuses on how molecular techniques are employed to identify bacterial species isolated from food, offering a detailed look at their application in microbiological analysis.ethod of using molecular methods to identify the species of isolated bacteria.
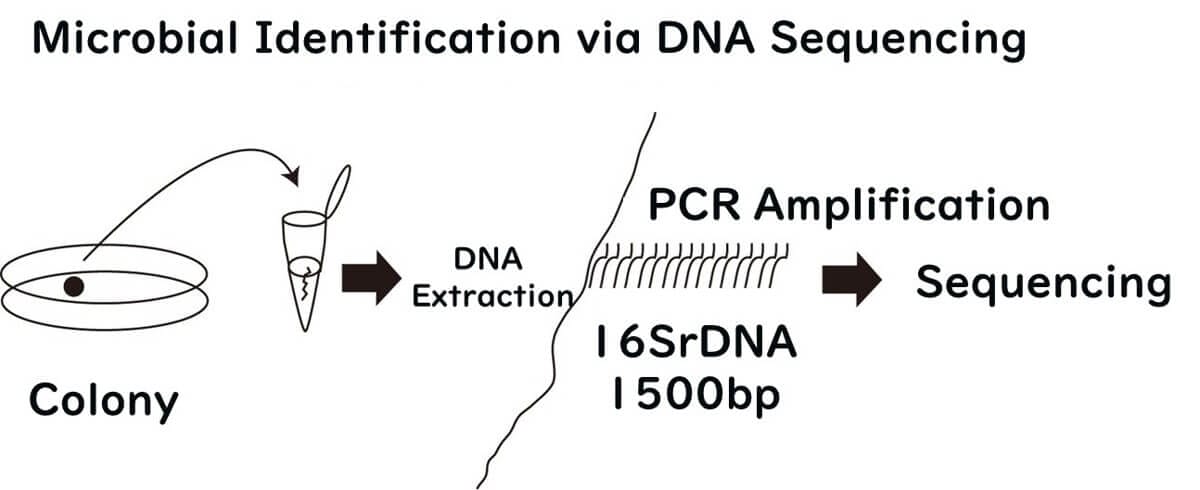
Identifying Bacteria Using Common DNA Sequences
Bacteria possess between 1,000 and 2,000 genes, depending on the species. To accurately identify bacterial species, it is essential to focus on DNA sequences that are universally present across all bacteria. These conserved sequences serve as reliable markers for species identification.
For example, Escherichia coli (E. coli) has a lactose-degrading enzyme gene, which is a distinguishing characteristic of the coliform group it belongs to. This gene is effective for identifying specific species within the coliform group. However, Salmonella lacks this enzyme, making the lactose-degrading gene unsuitable for identifying Salmonella species.
Housekeeping Genes: The Key to Accurate Species Identification
Housekeeping genes are a category of genes that all bacteria possess, and they are essential for the maintenance of life. The term "housekeeping" originates from the idea of fundamental tasks necessary for survival—akin to household chores that sustain daily life. Similarly, these genes perform critical functions that are indispensable for bacterial survival, making them universally present across all bacterial species.
Genes used for bacterial typing can be broadly divided into two categories: housekeeping genes and genes prone to variation. Housekeeping genes are involved in core survival functions, such as:
Ribosomal genes (e.g., 16S rDNA), essential for protein synthesis.
DNA replication genes (e.g., gyrB, coding for gyrase).
Stress response genes (e.g., heat shock proteins).
Mutations in housekeeping genes generally affect fundamental cellular processes. Bacteria with such mutations often fail to survive or are outcompeted, leading to a slow evolutionary rate for these genes. Moreover, environmental factors have minimal influence on whether these mutations are passed on, making housekeeping genes reliable as an "internal clock" of evolution.
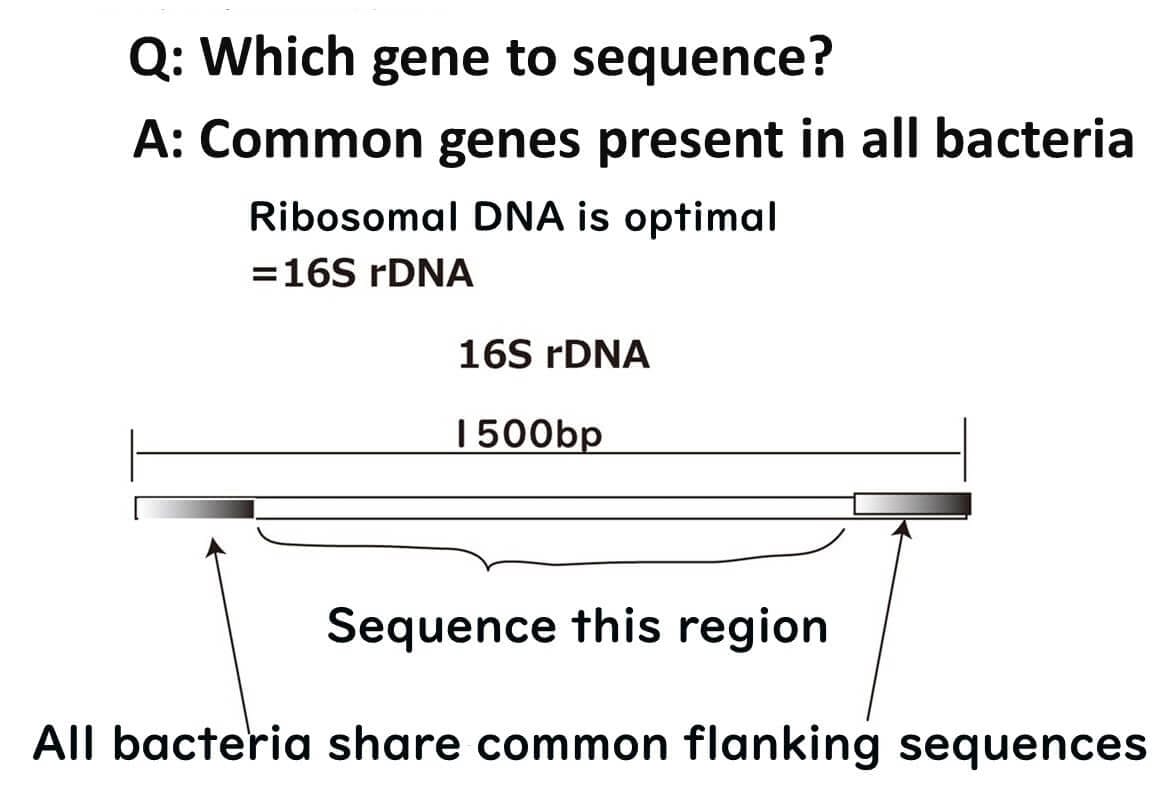
Among these, ribosomal RNA-related genes (e.g., 16S rDNA) stand out. Ribosomes, which are fundamental cellular organelles, are universally required for protein synthesis across all organisms. Therefore, 16S rDNA plays a pivotal role in bacterial species identification and remains a cornerstone in microbiological analysis.
Genes Unsuitable for Accurate Species Identification
While housekeeping genes are essential for bacterial survival, many other genes fall outside this category and are unsuitable for species identification. These include genes involved in non-essential functions, such as:
Metabolizing specific sugars.
Producing structures like flagella.
Determining serotypes or pathogenicity (except in obligate pathogens).
These genes are highly variable and prone to mutations influenced by environmental factors. Mutations in such genes are often passed down to offspring over long evolutionary periods, as long as they do not hinder survival.

From a phenotypic perspective, these variable genes may produce significant observable differences, such as the presence or absence of flagella, serotypes, or metabolic capabilities. This has historically led to the classification of closely related strains as entirely separate species. However, analysis based on housekeeping genes often reveals that these strains are part of the same species.
For example:
Photobacterium damselae (an opportunistic pathogen) and Pasteurella piscicida (a fish pathogen) were previously considered separate species due to differences in traits like flagella. However, 16S rDNA analysis integrated them into the same species.
Similarly, Escherichia coli (E. coli) and Shigella may differ in traits such as the presence of the lysine decarboxylase enzyme but are considered the same species at the molecular level.
This discrepancy underscores the importance of housekeeping genes, which mutate at a slow and consistent rate, making them a reliable "internal clock" for microbial evolution. In contrast, variable genes like those for lactose-degrading enzymes are heavily influenced by environmental factors, making them unsuitable for phylogenetic analysis.
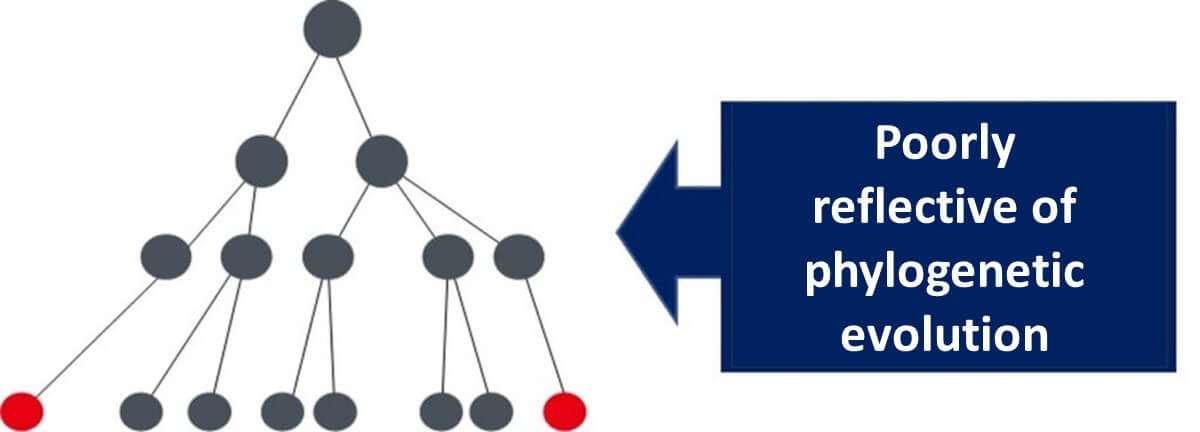
For instance, if E. coli inhabits an environment without lactose, it may lose its lactose-degrading enzyme gene. Conversely, other bacteria might acquire this gene via plasmids. Relying on such genes for evolutionary studies can lead to inaccurate phylogenetic trees.
Housekeeping Genes: Ideal Markers for Species Identification
Housekeeping genes, such as ribosomal RNA (rRNA) genes, are indispensable for cellular survival. Even minor mutations in these genes can render a cell nonviable, ensuring that only minimal genetic variations are passed down over generations. These mutations occur at an exceptionally slow rate, making housekeeping genes reliable indicators of evolutionary change.
For example, differences in the DNA sequences of bacterial 16S ribosomal RNA (16S rDNA) can be viewed as mutations that have accumulated gradually over the 3.3 billion years of bacterial evolution. These differences are invaluable for tracing microbial lineages and constructing accurate phylogenetic trees.
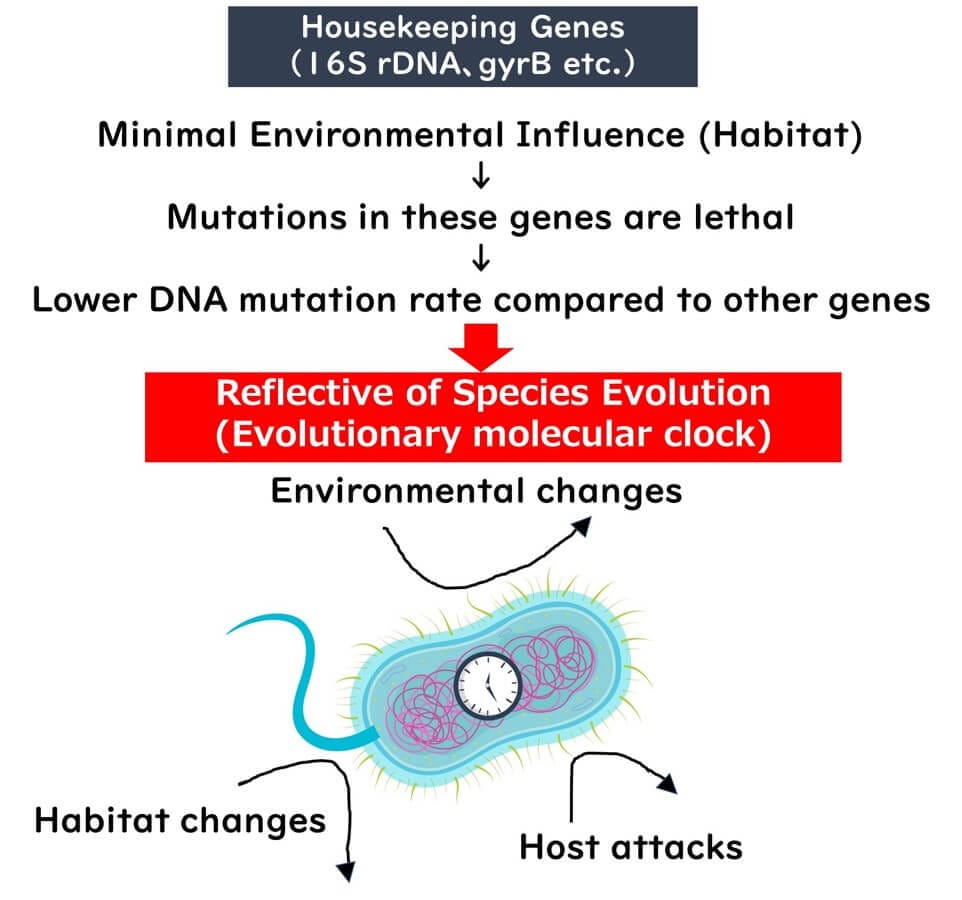
While mutations in housekeeping genes rarely persist due to their critical nature, certain regions within these genes are less essential. Mutations in these regions are not lethal and can accumulate over long evolutionary periods. This characteristic makes housekeeping genes ideal as an "internal clock" for studying evolutionary timelines.
In comparison to genes influenced by environmental factors, the evolutionary rate of housekeeping genes remains remarkably slow and consistent. As such, they provide a reliable framework for reflecting the true lineage of bacterial species.
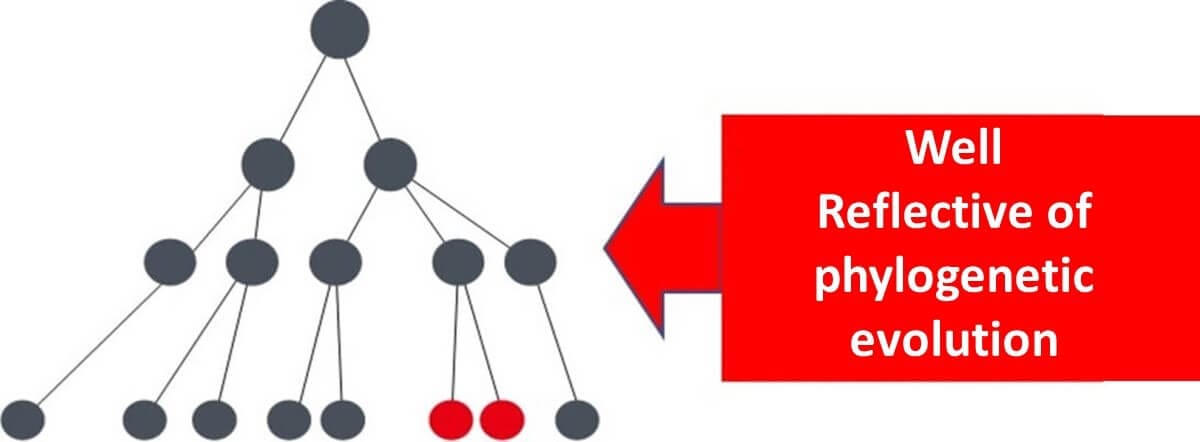
In summary, using housekeeping genes allows for the construction of phylogenetic trees that accurately represent microbial evolutionary history. Other genes, influenced by environmental variability, may fail to provide the same level of precision in reflecting evolutionary time.
Why Does the Ribosome Contain RNA?
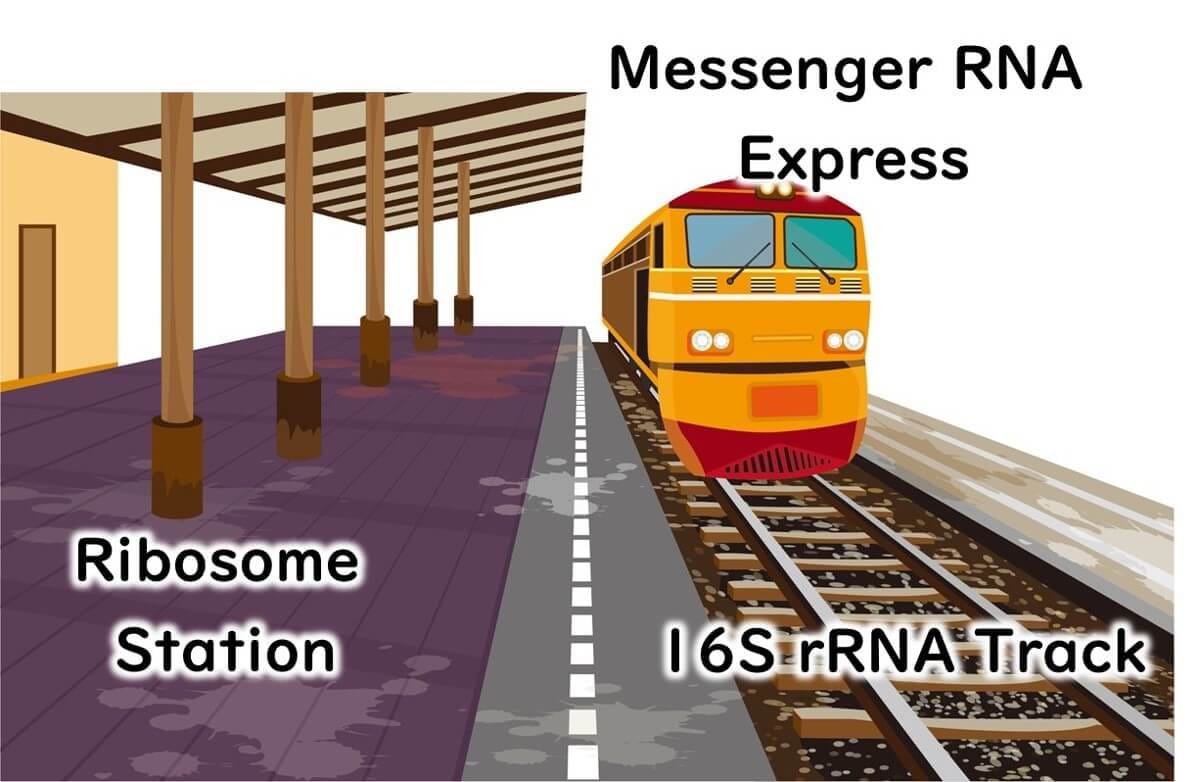
Ribosomes, the cell's protein synthesis machinery, are primarily composed of proteins, but they also contain RNA. Why is RNA essential in ribosomes? Messenger RNA (mRNA), transcribed from DNA, must bind to ribosomes for protein synthesis. This binding relies on the ribosomal RNA (rRNA) within the ribosomal proteins, which anchors the mRNA using the complementary base-pairing properties of nucleic acids.
To visualize this process, ribosomes can be likened to a train station, with mRNA acting as trains arriving at the platform. The tracks guiding the trains represent the ribosomal RNA, ensuring accurate alignment and interaction. Ribosomal RNA itself is transcribed from DNA, and for microbial identification, the DNA sequence encoding this RNA—known as 16S rDNA—is analyzed.
A brief note on the term '16S': The 'S' refers to the Svedberg unit, a measure of sedimentation rate during ultracentrifugation, which reflects the size of the molecule. Bacterial ribosomes are 70S, divided into 50S and 30S subunits. The 16S ribosomal RNA is part of the 30S subunit and plays a critical role in bacterial identification.
Mutation-Prone Regions in the 3D Structure of 16S rRNA
The 16S ribosomal RNA (16S rRNA) consists of approximately 1500 nucleotides, forming a three-dimensional structure with two distinct regions: stem-like structures and loop regions.
Stem-like structures: These regions are stabilized by complementary base pairing. Mutations in the DNA sequence encoding these regions significantly disrupt the overall 3D structure of 16S rRNA, often resulting in cell death. Consequently, mutations in these regions are rare and unlikely to persist over evolutionary time.
Loop regions: These single-stranded areas are not involved in base pairing and are less structurally critical. Mutations here do not impact the ribosomal RNA's overall function and are more likely to accumulate over time. These mutations are passed down through generations, reflecting the evolutionary history of bacteria.
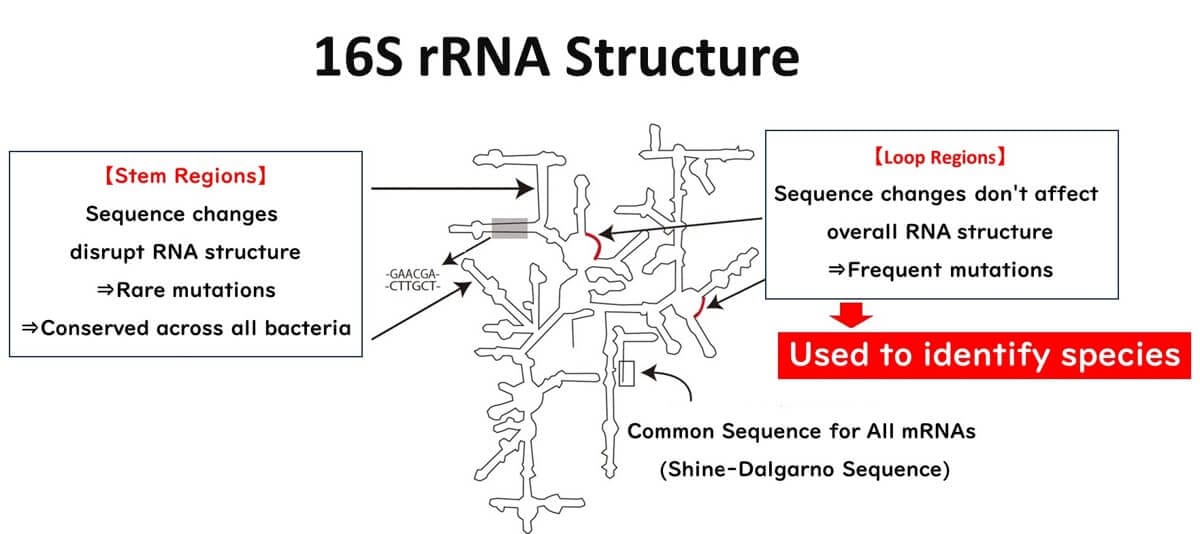
Because of this distinction, the loop regions serve as a reliable "internal clock" for evolutionary studies, as they accumulate mutations spontaneously, independent of environmental pressures. This property makes them particularly suitable for species identification.
When analyzing 16S rRNA for species identification, examining all 1500 nucleotides can be experimentally challenging. Instead, focusing on the variable loop regions, such as the V3-V4 regions or the first 500 base pairs, provides an efficient and accurate method for identifying bacterial species.

16S rDNA Identification: More Than Just an OTU
In food microbiology, identifying microbes often involves determining bacterial "species." However, the relationship between genetic sequences and species classification is not always straightforward.
Unlike higher plants, bacteria lack consistent and reliable morphological features for classification. There is no universally agreed-upon definition of what constitutes a "species" in bacteria. Bacterial reproduction through binary fission enables rapid genetic mutations, resulting in groups of clones with slightly different genetic sequences. Defining species boundaries within such groups is inherently complex.
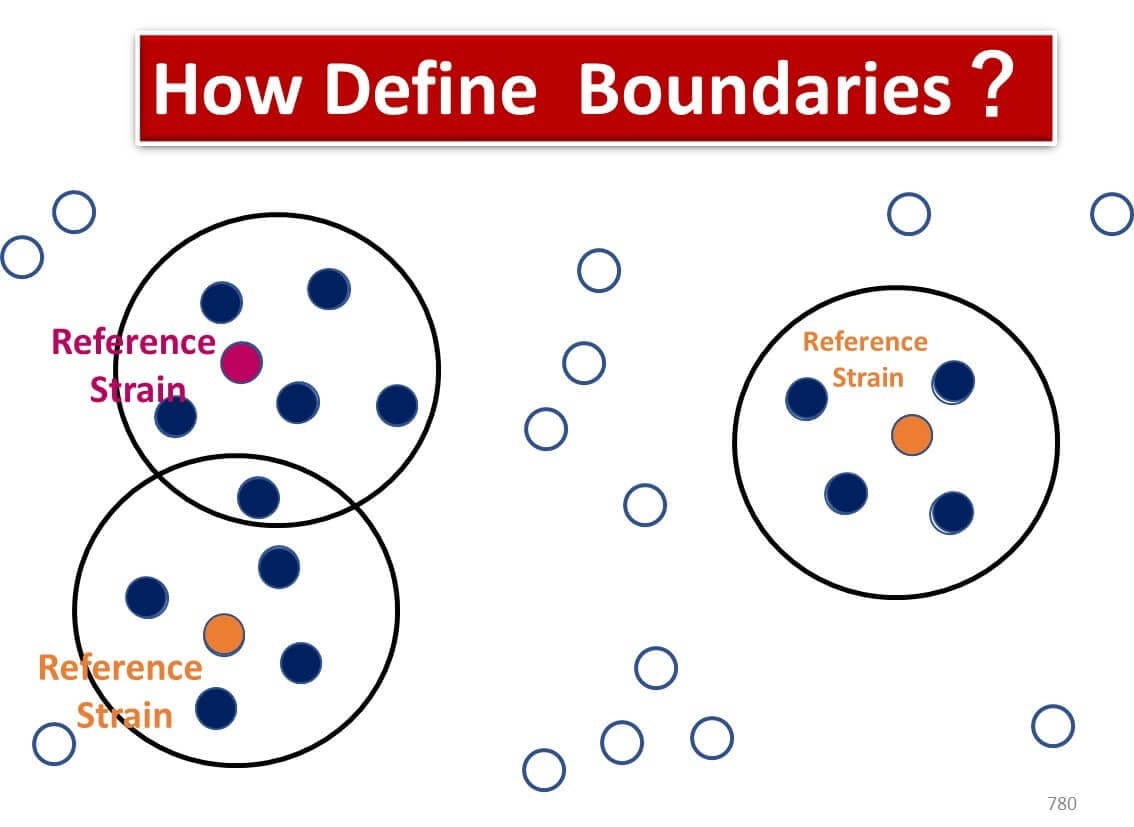
Currently, bacterial species are classified using a combination of phenotypic traits, including:
Gram staining.
Morphology.
Physiological and biochemical characteristics.
Since the 1990s, advancements in bacterial genomics have introduced DNA-DNA hybridization as a key standard. Bacteria are defined as the same species if their DNA-DNA hybridization rate exceeds 70% with a type strain. This method forms the foundation of species identification based on genetic homology.
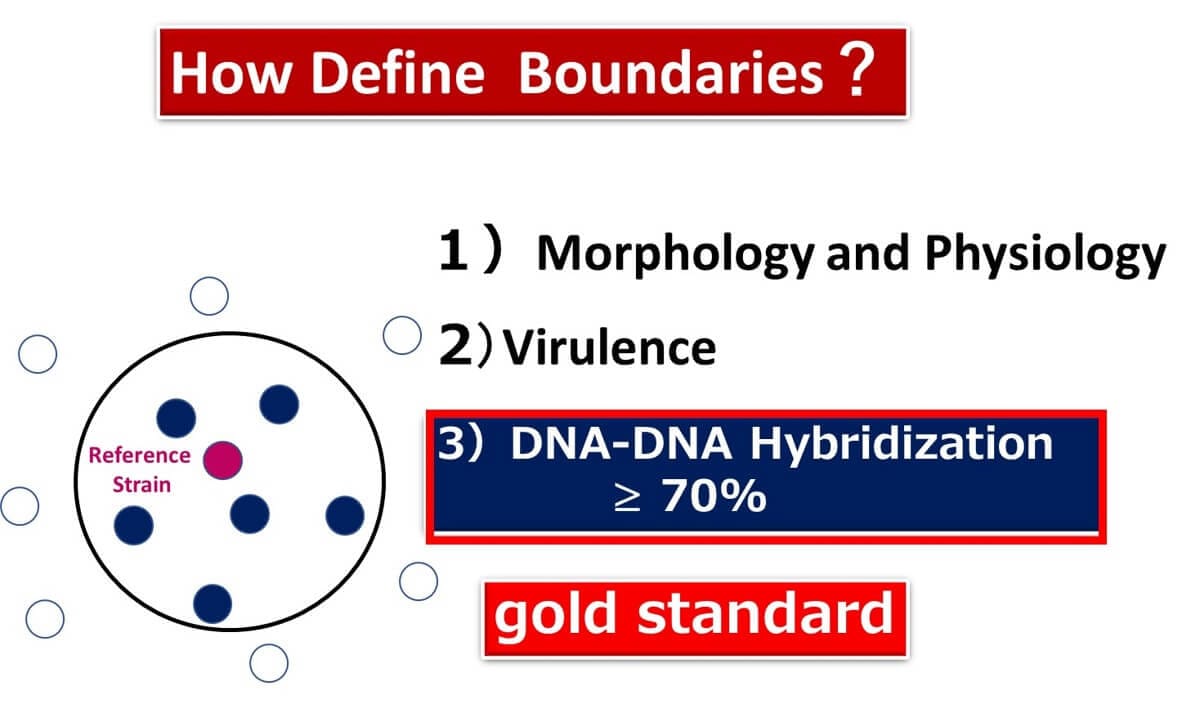
What Does 16S rDNA Identification Offer?
Dr. Carl Woese revolutionized microbiology in 1977 by introducing 16S rDNA for phylogenetic analysis. Since then, 16S rDNA analysis has become an indispensable tool for microbial classification and ecological studies. Unlike DNA-DNA hybridization, which is technically demanding and time-consuming, 16S rDNA analysis offers a practical alternative.
Based on personal experience in the 1990s, I found DNA-DNA hybridization labor-intensive, requiring over a month of practice to achieve reliable results. The advent of 16S rDNA-based identification was a game-changer, simplifying bacterial classification processes.
The Concept of OTU: Operational Taxonomic Unit
A crucial concept in 16S rDNA-based identification is the Operational Taxonomic Unit (OTU). An OTU is a computationally defined unit, representing groups of bacterial sequences with similarity above a chosen threshold. Importantly, OTUs are not true species but convenient taxonomic units for classification. In microbial ecology and taxonomy, OTUs derived from 16S rDNA sequences are often treated as equivalent to species for practical purposes.
While full-length 16S rDNA analysis provides comprehensive insights, the "species" identified through this method are more accurately described as OTUs rather than biological species. This distinction is essential for understanding the limitations and utility of 16S rDNA-based methods in microbial identification.

The Limitations of 16S rDNA Identification
Since the 1990s, 16S rDNA analysis has become a widely used tool for bacterial identification. Initially, taxonomists agreed that if the full-length 1500 base pairs of 16S rDNA showed over 97% similarity, the organisms were likely the same species. This threshold correlated with DNA-DNA hybridization rates of 70% or more, which was the standard for defining bacterial species.
However, this 97% threshold has proven inadequate for some bacterial species. Recent studies suggest that a higher similarity threshold—98.7% or even 99%—is more accurate for defining species. For example:
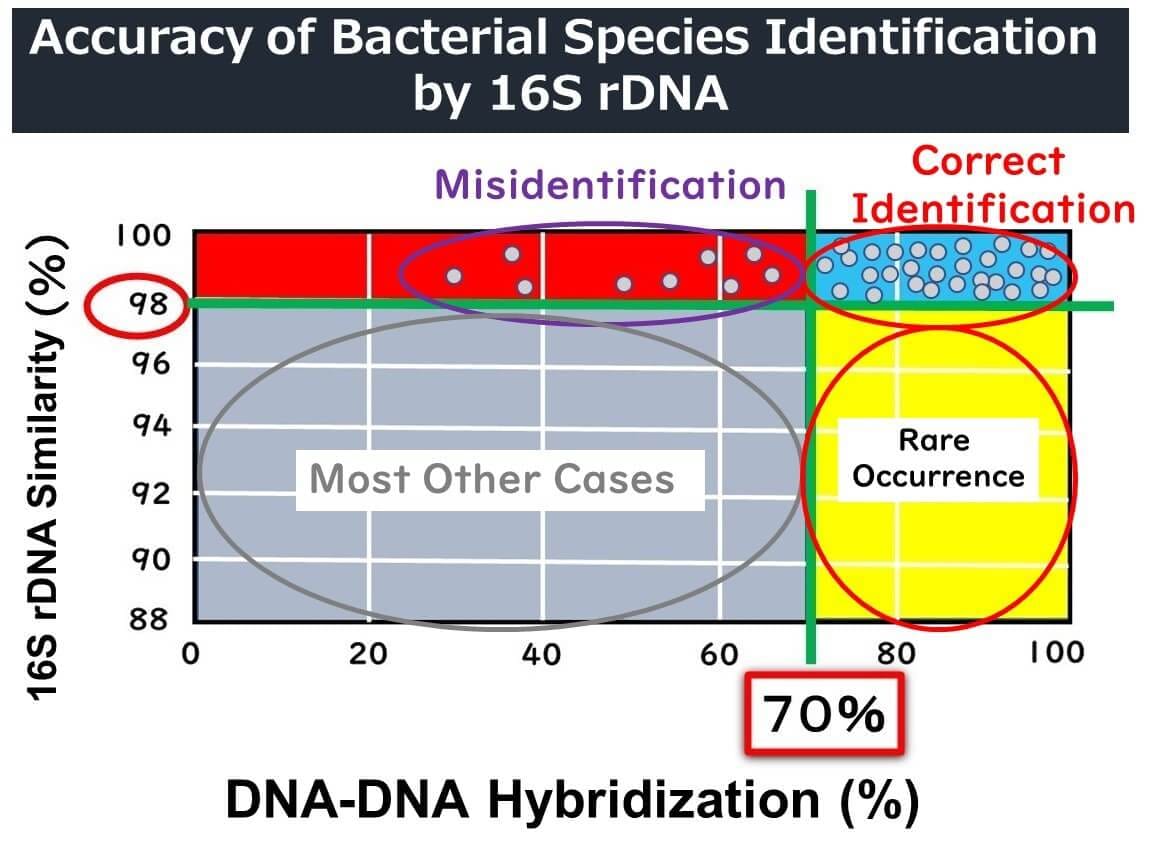
Misidentifications and the Challenge of Thresholds
Even when 16S rDNA sequences show 98% similarity, DNA-DNA hybridization rates can fall below the 70% benchmark, indicating that the organisms might not be the same species. Conversely, organisms with less than 98% 16S rDNA similarity are almost never the same species. While this provides some guidance, relying solely on 16S rDNA similarity can lead to misidentifications.
For example:
OTUs classified as the same species based on 16S rDNA may include cocci and bacilli, mixtures of Gram-positive and Gram-negative bacteria, or organisms with different oxygen requirements.
Such inconsistencies highlight the limitations of 16S rDNA as a sole species identification tool.

Practical Implications for Food Microbiology
In practical terms, species estimation based on 16S rDNA is often sufficient for routine bacterial testing in food microbiology. However, it is essential to recognize its limitations. Full species identification requires considering phenotypic traits, such as morphology and biochemical characteristics, or using DNA-DNA hybridization as outlined in Bergey's Manual of Systematic Bacteriology.
For food companies, where routine testing prioritizes efficiency, 16S rDNA offers a practical and reliable method for species estimation. However, microbiologists must remain cautious and aware of its potential for misclassification.

Related posts:
DNA Testing vs. Culturing in Microbiology: Speed, Accuracy, and Global Data Compatibility (3) Unveil the magic of genetic testing in microbiology—a swift and...

Discover expert-led lessons in food microbiology designed for professionals and beginners alike.

Author of this Blog: Bon Kimura
Bon Kimura, Professor Emeritus at Tokyo University of Marine Science and Technology (TUMSAT), specializes in food microbiology. He obtained his PhD from Kyoto University and became a professor at TUMSAT in 2006, serving as Dean of the Faculty of Marine Science from 2012-2015. Kimura has published over 200 international papers on food safety, pathogens, and spoilage bacteria. He has received multiple awards, including the Japanese Society for Food Microbiology Award (2019). Kimura also served as an editor for the International Journal of Food Microbiology from 2012 to 2024, where he was the principal reviewer for 1,927 papers.
Copyright © Introduction to Food Microbiology and Safety All Rights Reserved.