Introduction to Food Microbiology and Safety
Bacterial Strain Typing: PFGE, MLST, and MLVA in Molecular Epidemiology
Bacterial strain typing plays a crucial role in identifying and tracking foodborne pathogens and outbreaks. Before whole-genome sequencing (WGS) became standard, traditional molecular epidemiology methods like PFGE, MLST, and MLVA were the primary tools for analyzing bacterial strains and genetic relationships.. Among these, Pulsed-Field Gel Electrophoresis (PFGE), Multi-Locus Sequence Typing (MLST), and Multiple-Locus Variable-number Tandem Repeat Analysis (MLVA) played a crucial role in identifying bacterial strains and understanding their genetic relationships.
This article provides an in-depth look at these traditional molecular epidemiology methods, explaining how they have been used in food safety and outbreak investigations.
The Importance of Strain Identification
Identifying bacterial strains is crucial in molecular epidemiology and foodborne outbreak investigations. When a food poisoning incident occurs, public health agencies conduct molecular epidemiological analysis to determine the causative bacterial strain. However, simply identifying the species using 16S rRNA gene (16S rDNA) sequencing is insufficient.
16S rDNA Sequencing: Species Identification, Not Strain Differentiation
Imagine trying to determine whether an unknown creature is a fox, a monkey, or a human. Using 16S rDNA sequencing, you can classify the organism at the species level. However, this method cannot differentiate between individual members of the same species.
For those interested in the basics of bacterial species identification through genetic analysis, please refer to the article below:
Streamlined Molecular Methods for Microbial Identification: A Practical Guide
Why Strain Identification Matters: The Alien Analogy
Now, imagine a scenario where an alien spacecraft orbiting Earth is shot down by someone on the planet.

The alien captain wants to find out who is responsible.
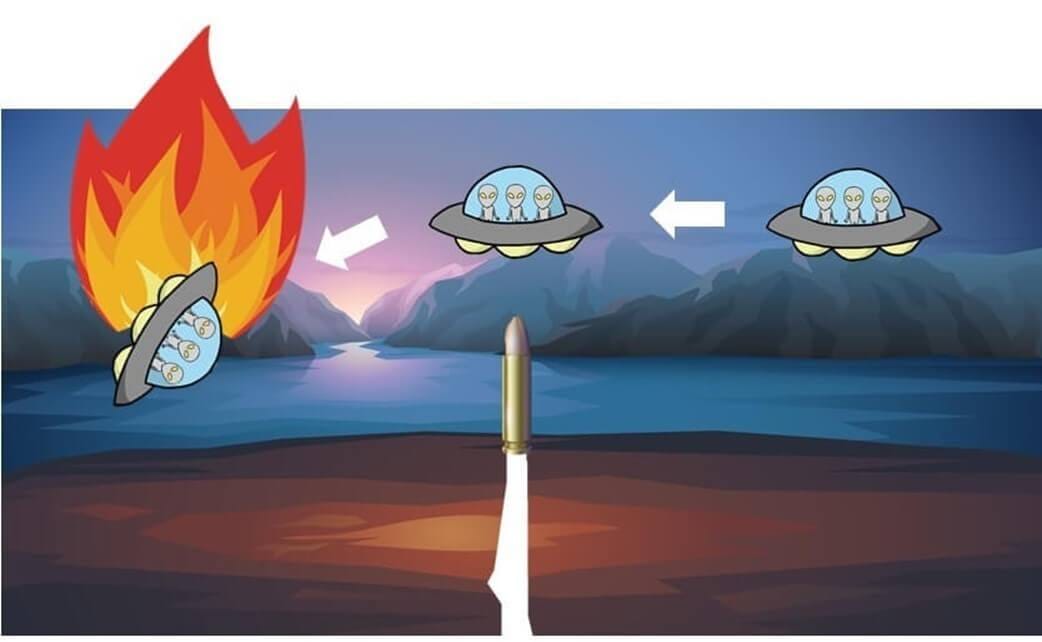
If the aliens only know that the culprit is a Homo sapiens, they might feel compelled to destroy all humans on Earth. However, the actual culprit is a specific individual within the Homo sapiens species.

In the same way, molecular epidemiology must go beyond species identification to pinpoint specific bacterial strains responsible for outbreaks.
Using strain-level identification, public health agencies can accurately trace bacterial transmission routes, just as the aliens could identify and arrest only the specific individual responsible instead of punishing all humans.

Whole-Genome Sequencing (WGS) vs. Traditional Strain Typing: Pros and Cons of Both Methods
Identifying bacterial strains requires a much higher resolution than species-level identification. While 16S rRNA gene (16S rDNA) sequencing can classify bacteria at the species level using approximately 1,500 base pairs, this limited genetic information is insufficient for strain differentiation.
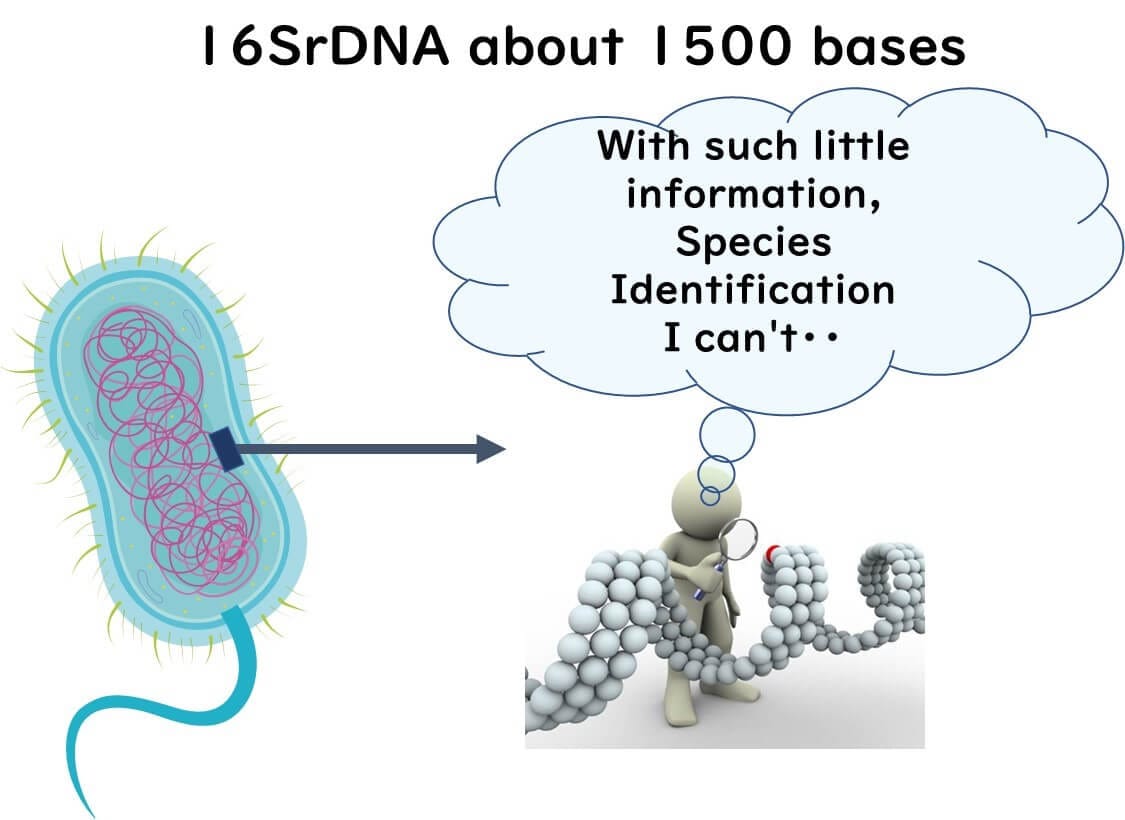
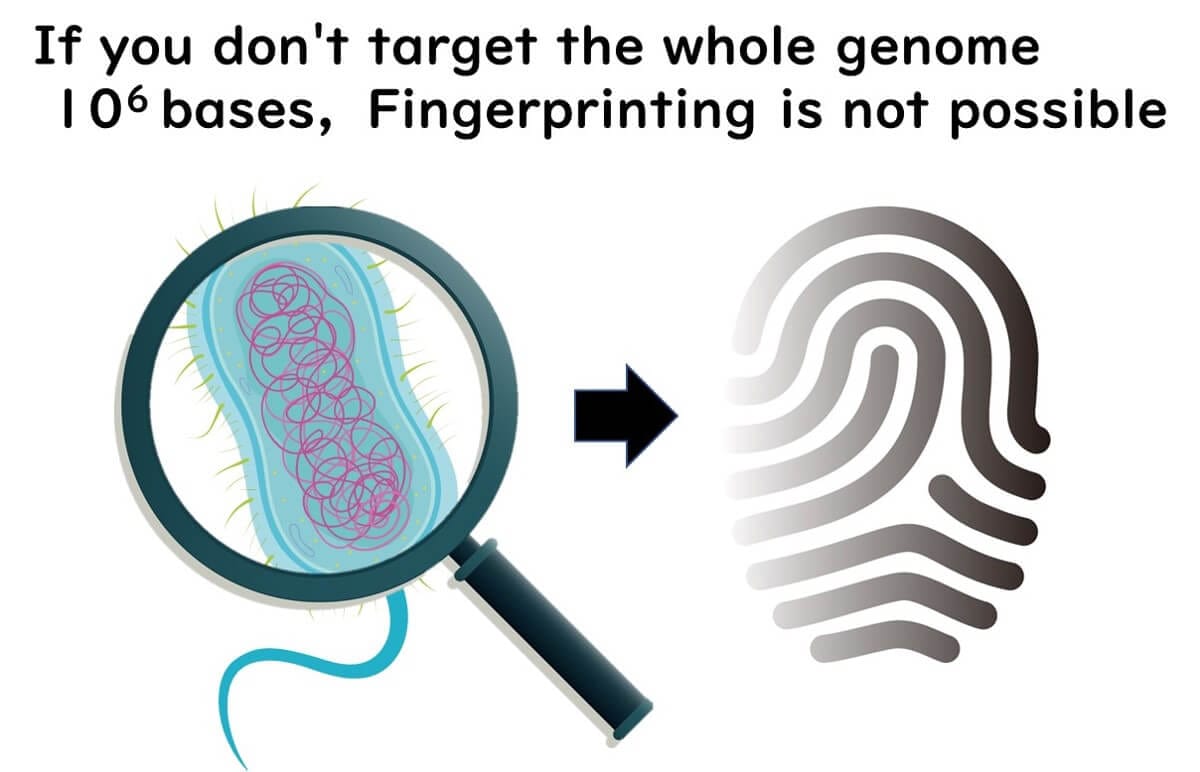
Why Whole Genome Data is Essential for Strain Typing
The entire bacterial genome consists of approximately 10⁶ base pairs. Without analyzing the whole genome, precise strain typing—also known as fingerprinting—is not possible.
To accurately differentiate bacterial strains, molecular epidemiology relies on methods that examine the entire genome rather than just a single gene region. This higher resolution allows public health agencies to trace infection sources, track outbreaks, and implement targeted interventions.
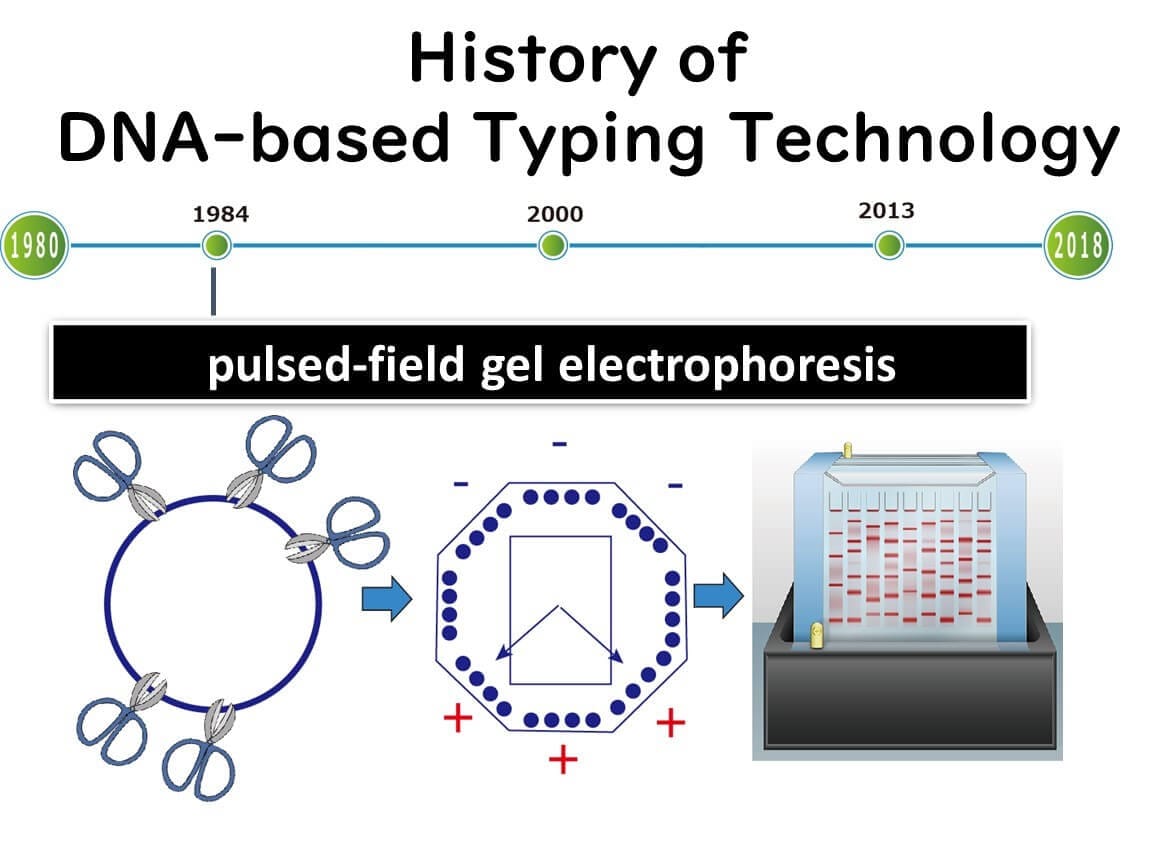
Pulsed-Field Gel Electrophoresis (PFGE)
The Rise of PFGE in the 1990s
Introduction to PFGE
Pulsed-field gel electrophoresis (PFGE), introduced in the 1990s, became the primary method for bacterial molecular epidemiology until around 2010. It was widely used to differentiate bacterial strains in foodborne outbreak investigations, allowing public health agencies to track pathogen transmission more precisely.
What is PFGE?
PFGE is a fragment analysis technique that identifies bacterial strains by cutting the genome with restriction enzymes and analyzing DNA fragment patterns through electrophoresis. Ideally, comparing entire genomes provides the most accurate strain identification, but in the 1980s, sequencing even small portions of bacterial genomes was technically challenging.
Thus, PFGE emerged as one of the few practical high-resolution molecular typing methods from the 1990s onwards. Even today, it remains in use in many countries, although the United States ceased PFGE-based surveillance in 2019 in favor of whole-genome sequencing (WGS).
How PFGE Works
PFGE uses restriction enzymes to cut bacterial DNA at specific sequences, generating large DNA fragments. The fragments are then separated by electrophoresis, but unlike conventional methods, PFGE alternates the electric field direction at set pulse times. This technique allows large DNA fragments (around 10⁴ bp) to migrate through the gel, enabling better resolution for strain differentiation.
For example, when cutting smaller DNA fragments, such as 16S rDNA (~1,500 bp), standard electrophoresis with a direct current is sufficient. However, large DNA fragments require PFGE's alternating electric field to move through the gel efficiently.
For an overview of electrophoresis techniques, see:
What is PCR? Basics, Process, and Applications in Food Microbiology Testing
PFGE in Molecular Epidemiology
PFGE became the gold standard for bacterial strain typing due to its ability to detect genetic variations based on fragment patterns. In 1996, the PulseNet network, led by the CDC, was established to standardize PFGE protocols and create a global database for foodborne outbreak investigations.
Because PFGE examines the entire genome, it offers much higher resolution than single-gene sequencing methods like 16S rDNA (which typically examines only 1,000–2,000 bases).
However, with the advent of next-generation sequencing (NGS) after 2010, whole-genome approaches have largely replaced PFGE, especially in high-throughput public health laboratories.
Fragment Analysis vs. DNA Sequencing: Why is Fragment Analysis Declining?
While PFGE has played a crucial role in bacterial strain typing, it is a fragment analysis technique that comes with several drawbacks. These limitations impact data precision, reproducibility, and global standardization, which has led to its gradual replacement by whole-genome sequencing (WGS) in molecular epidemiology.
Identification Precision and Data Compatibility
All fragment analysis techniques, including PFGE, rely on the number and mobility of genetic fragments on an electrophoresis gel. However, there are several challenges associated with this approach:
Subjectivity in Band Interpretation: Variability in fragment inclusion criteria, band intensity thresholds, and minor mobility differences between laboratories can lead to inconsistencies.
Limited Genetic Information: Fragment analysis only examines the molecular size of gene fragments, not their exact sequence. This can cause sequence variations within fragments of the same size to be overlooked.
Low International Compatibility: The PulseNet database, which standardizes PFGE worldwide, requires complex standardization efforts due to the variability in fragment analysis techniques.
Not Suitable for Phylogenetic Analysis: Since fragment analysis does not directly compare genetic sequences, it is not ideal for determining evolutionary relationships between bacterial strains.
Key Takeaway: PFGE-based strain typing has high resolution, but lacks precision at the sequence level, making data sharing across laboratories challenging.

Practical Limitations and Cost Issues
Another disadvantage of PFGE is its complexity and time-consuming nature. The experimental process is labor-intensive, often requiring several days to obtain results.
To address these issues, some private food companies have adopted alternative fragment analysis methods such as:
AFLP (Amplified Fragment Length Polymorphism)
Ribotyping
While these methods offer some advantages, they also come with high costs and technical complexity, making them less accessible for routine public health surveillance.
The Decline of Fragment Analysis in Favor of Sequencing
Historically, fragment analysis methods dominated bacterial strain identification because DNA sequencing technology was expensive and slow. In the early 1990s, PCR-based 16S rDNA sequencing was introduced, but due to technological limitations, only 1,500 bases could be read at a time.
To work around these limitations, restriction enzyme-based methods such as RFLP (Restriction Fragment Length Polymorphism) were used to classify bacteria based on fragment patterns rather than gene sequences. This approach was widely used in the 1990s but became obsolete with the advent of capillary sequencers in the 2000s, which made DNA sequencing cheaper and faster.
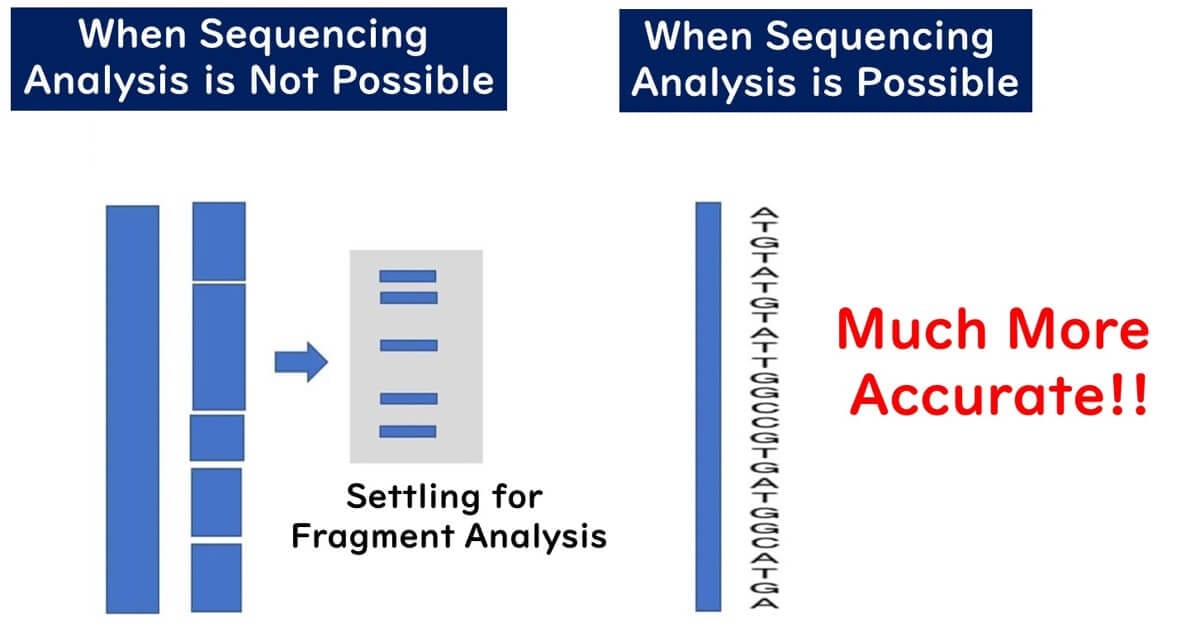
MLST vs. MLVA: Key Differences Between DNA-Based Molecular Epidemiology Methods
With the rapid spread of capillary sequencers in the 2000s, the pace and cost of DNA analysis decreased significantly. This advancement made it possible to routinely analyze multiple gene regions, typically 5-6 genes, with each consisting of several hundred bases, for numerous bacterial strains. This capability led to the development of Multilocus Sequence Typing (MLST). Since its development in 1998 and widespread adoption in 2000, MLST has been widely used for analyzing various foodborne and pathogenic bacteria.
Multilocus Sequence Typing (MLST) Method
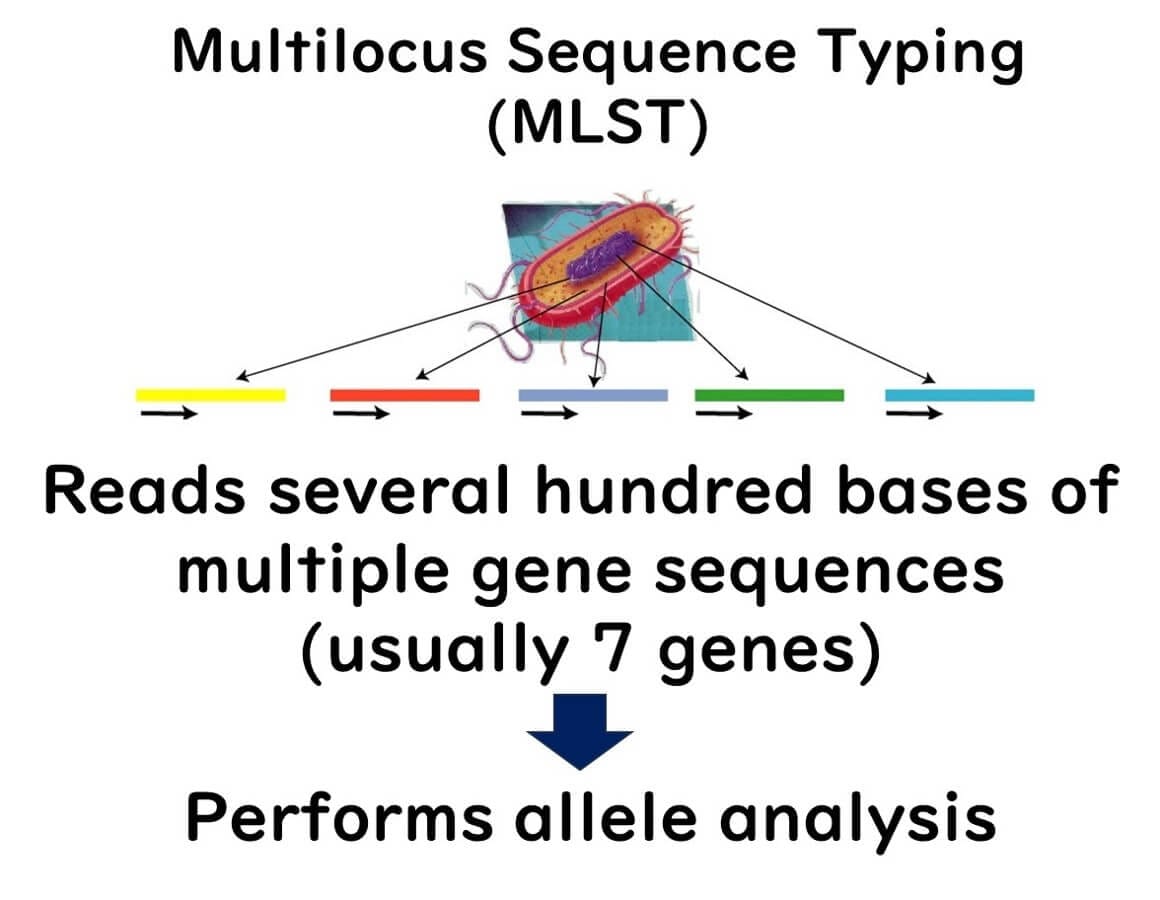
MLST involves selecting seven genes from a microorganism's genome and sequencing them. By 2000, reading partial sequences of seven genes from microorganisms became feasible for a typical public research laboratory.

This method involves sequencing about 400 bases from multiple gene regions (usually seven or more) and using these sequences for bacterial typing. Each strain's differences in multiple gene sequences are categorized into alleles for classification, and comprehensive analysis with integrated genetic analysis software achieves higher-resolution strain identification than PFGE.
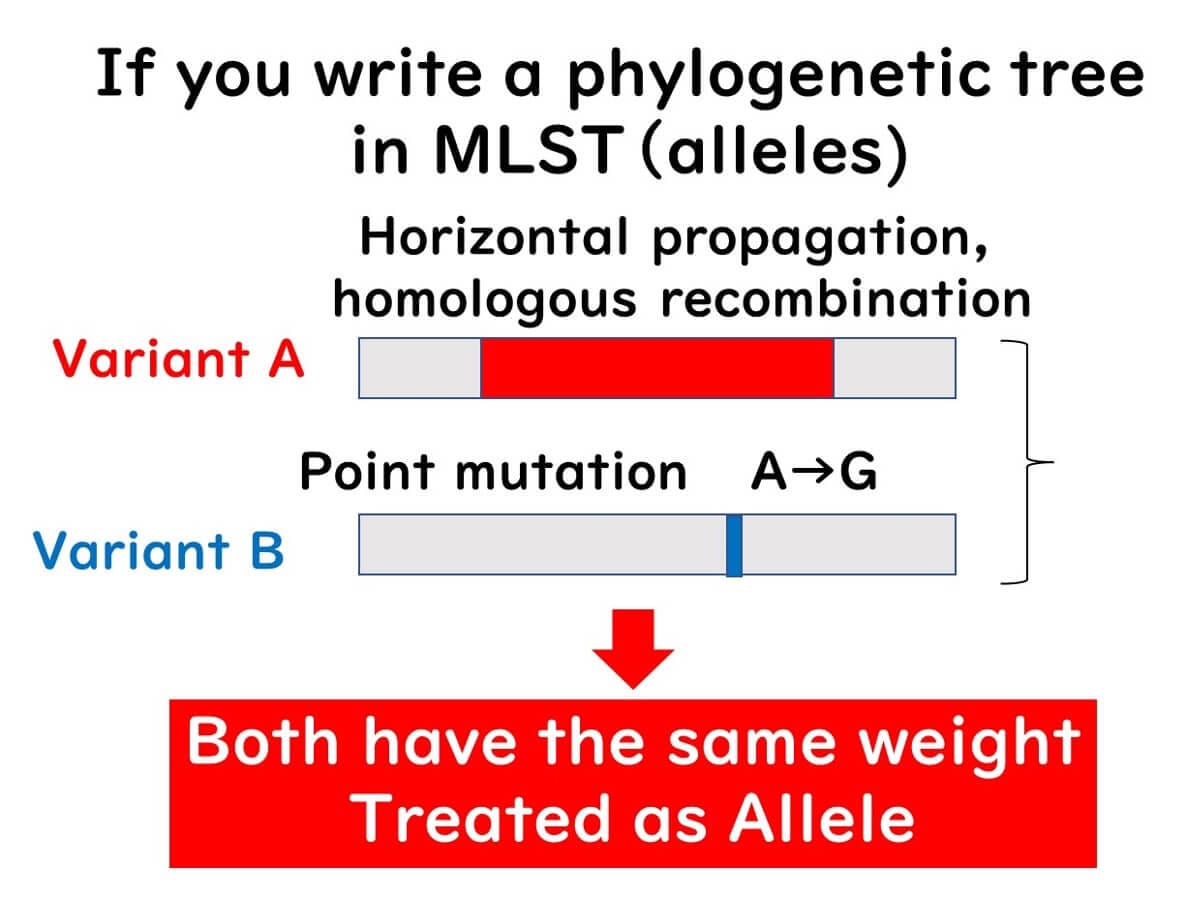
Unlike traditional phylogenetic trees based on gene sequences, MLST groups the differences in gene sequences and constructs phylogenetic trees based on allelic profiles. For example, if there are genetic differences between strains A, B, and C, they are classified into different alleles.
MLST Analysis
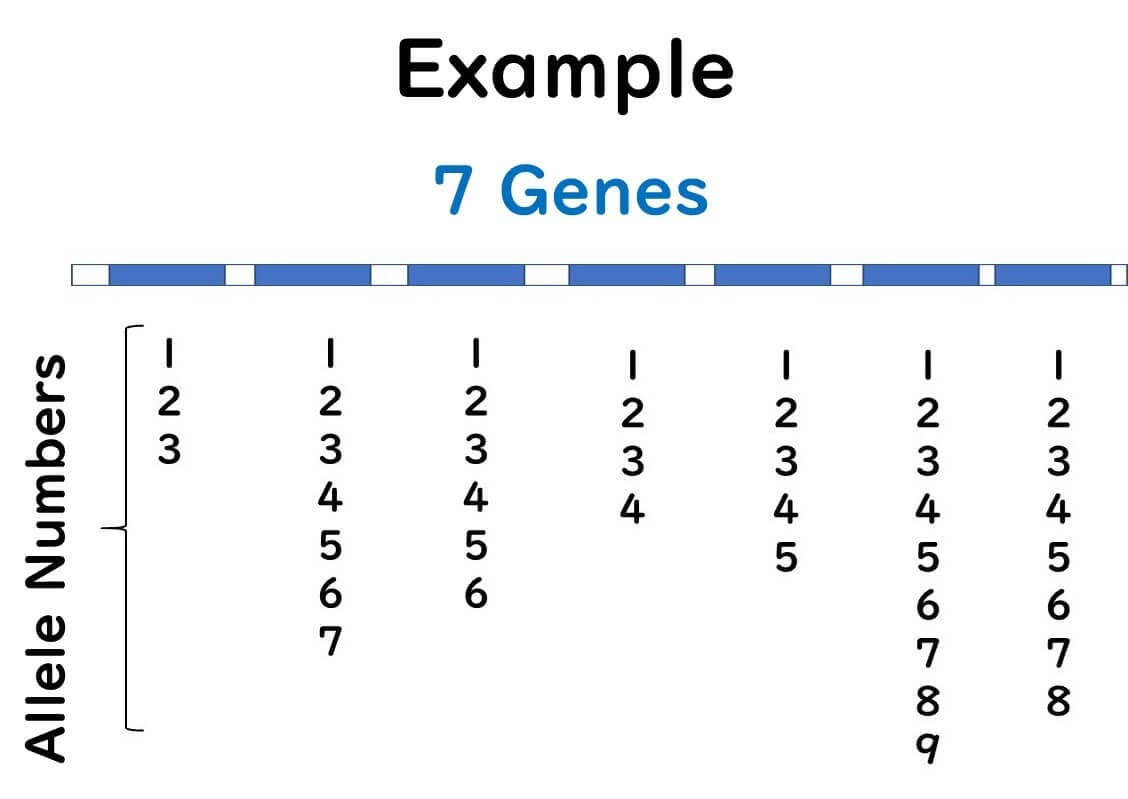
Within a single gene, even a single base difference results in a different allele number.
Regardless of how many sequence differences exist, distinct alleles are assigned based on any variation.

Unlike traditional methods, where the degree of sequence variation determines classification, MLST treats differences between strains A, B, and C equally by assigning distinct allele numbers.
Advantages of MLST
The primary advantage of MLST is that it is based on genetic sequence information, resulting in digital outputs. PFGE, relying on visual detection of band positions, can introduce subjectivity. MLST analysis, based on genetic sequence information, eliminates ambiguity and allows for easy comparison of data across laboratories. This makes it ideal for database registration and sharing strain data among multiple research institutions. Genetic sequence information, such as DNA sequences, can be analyzed consistently and accurately by any lab worldwide, enabling precise and common data exchange over the internet.
While species-level identification using specific gene sequences (e.g., 16S rDNA, gyrB) has been done, strain-level identification requires more than one or two gene sequences (usually around 1,000 bases). Therefore, strain identification using genetic sequence information had not been feasible until MLST.
Additionally, MLST, unlike PFGE, involves simple operations to determine genetic sequences. Subsequent sequence editing and phylogenetic analysis are facilitated by available commercial software, making the process straightforward. Furthermore, multi-capillary sequencers automate much of the procedure.
Why Use Multiple Gene Sequences (Multi Locus)?
Let's delve into the theoretical background of the MLST method. As mentioned earlier, the emergence of MLST was a natural progression driven by the rapid advancements in DNA analysis speed and cost reduction. However, another critical factor is the significant shift in our understanding of bacterial phylogeny over the past 20 years.
Our perception of bacterial genome evolution has changed from viewing bacterial populations as clonal to recognizing them as non-clonal populations. In the 1990s, when researchers could read bacterial gene sequences (often 16S rDNA), it was believed that bacterial gene evolution primarily occurred through point mutations passed down to descendants. However, since 2000, as various bacterial gene sequences have been uncovered worldwide, it has become clear that bacterial gene evolution frequently involves homologous recombination of large gene fragments, far more common than previously imagined.
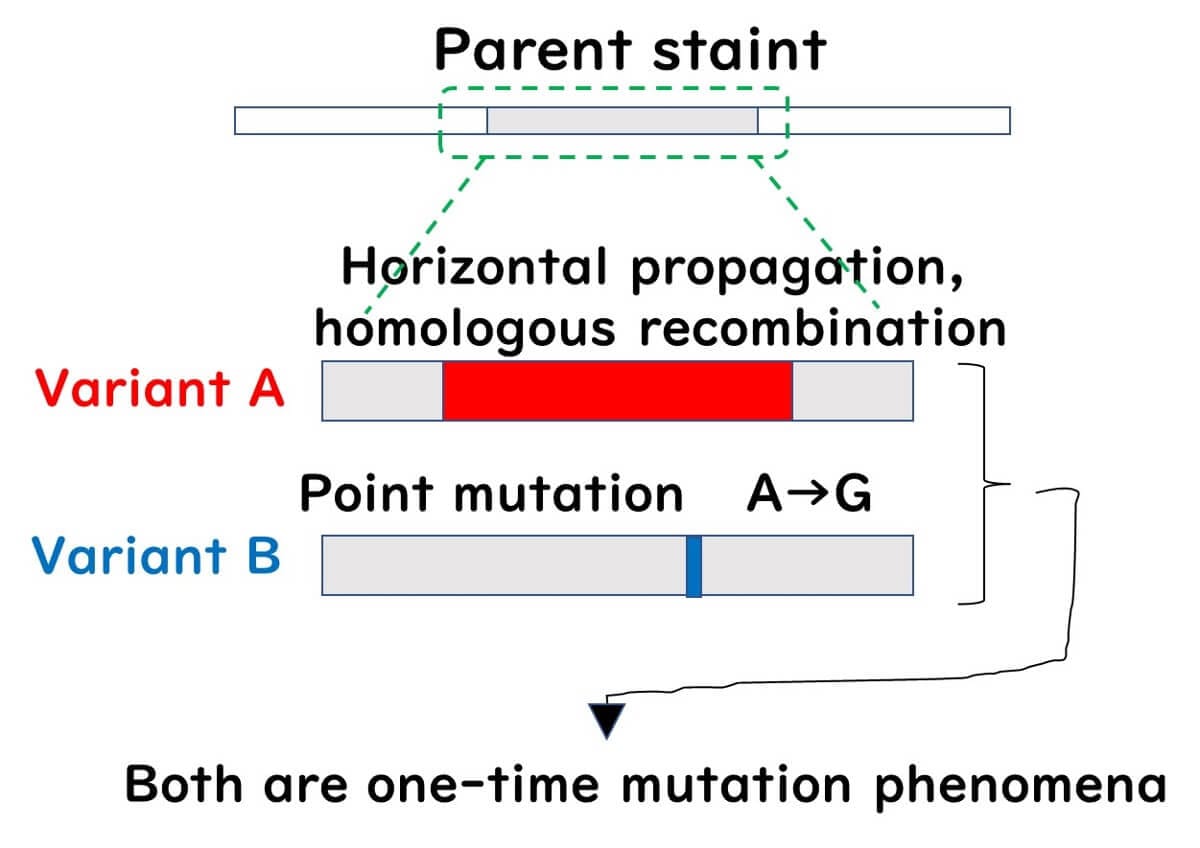
When bacterial gene evolution is seen solely as the accumulation of point mutations passed down to descendants, we describe the bacterial population as a clonal population. Conversely, a population with frequent exchanges of large gene fragments through homologous recombination is described as a non-clonal population.
In clonal populations, a phylogenetic tree based on the sequence of a single gene can accurately reflect the phylogeny. However, in non-clonal populations, a phylogenetic tree based on a single gene can be misleading if a single gene has undergone significant homologous recombination, placing the recombinant strain far from its true phylogenetic position. Meanwhile, strains with multiple point mutations but no recombination might appear closer to the original strain than they should be.
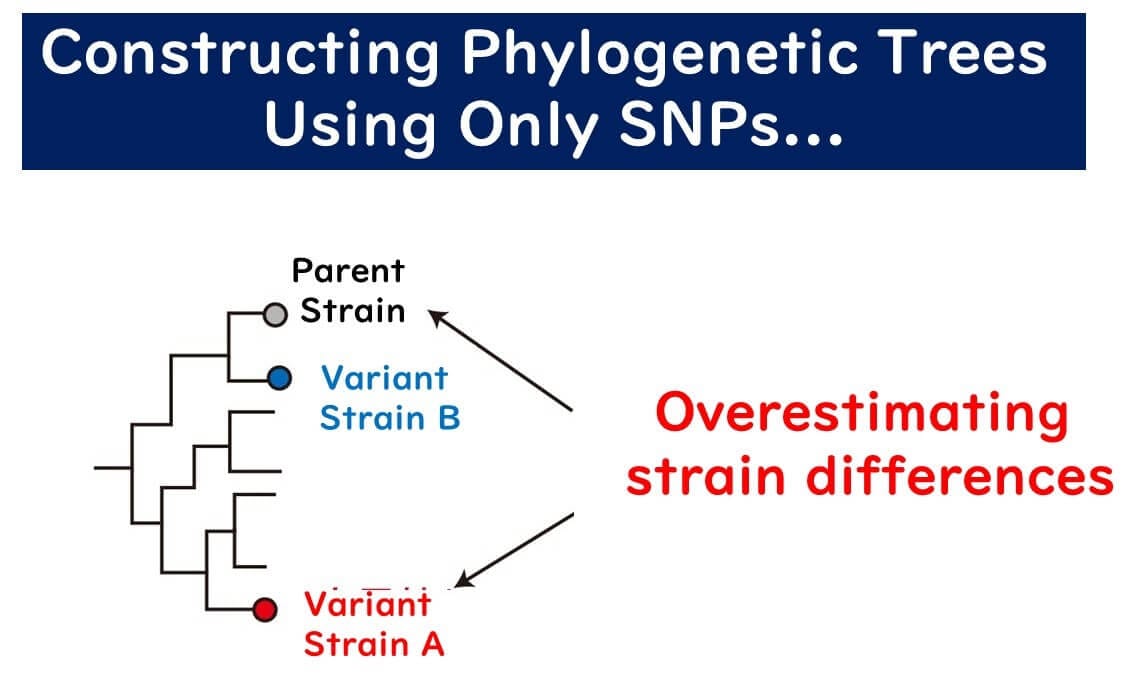
In non-clonal populations, constructing a phylogenetic tree from a single gene is likely to produce errors. MLST addresses this by:
Using multiple gene sequence information to construct phylogenies.
Constructing phylogenies based on differences in gene sequences (alleles) rather than the sequences themselves.
This approach allows MLST to accurately reflect the evolutionary timeline of non-clonal populations.

Which Genes Are Used in MLST Analysis?
Next, let's discuss the genes used in MLST analysis. Genes used in bacterial typing can be broadly divided into housekeeping genes and other rapidly evolving genes. Housekeeping genes are crucial for bacterial survival, such as ribosomal genes (16S rDNA), gyrase genes involved in DNA replication (gyrB), and stress-related genes, such as heat shock protein genes.
Mutations in housekeeping genes significantly affect the cell's essential functions, causing most mutated cells to die or be eliminated from the population. Consequently, the evolutionary rate of these genes is slow, and their evolution is not significantly influenced by environmental factors, acting as a "molecular clock" for evolutionary studies. In MLST, using housekeeping genes is the fundamental approach.
For more on the evolutionary compatibility of housekeeping genes and other genes, see the article:
MLVA Method
As discussed in the section on MLST, pulsed-field gel electrophoresis (PFGE) has limitations in terms of procedural complexity and data objectivity for bacterial strain typing. To address these issues, MLVA (Multiple-Locus Variable-Number Tandem-Repeat Analysis) was developed, focusing on repetitive sequences in bacterial genomes for strain identification. MLVA was introduced slightly after MLST, around the year 2000, and has since become a valuable tool.
MLVA determines the number of repetitive DNA sequences (tandem repeats) dispersed throughout the bacterial genome. The number of these tandem repeats at specific loci varies between bacterial strains. These loci-specific repeat numbers are known as VNTRs (Variable-Number Tandem Repeats), which exist at multiple loci or regions within the genome.
Research has demonstrated that VNTRs are useful for strain-level identification of bacterial strains. MLVA is particularly suitable for highly clonal organisms like E. coli O157.
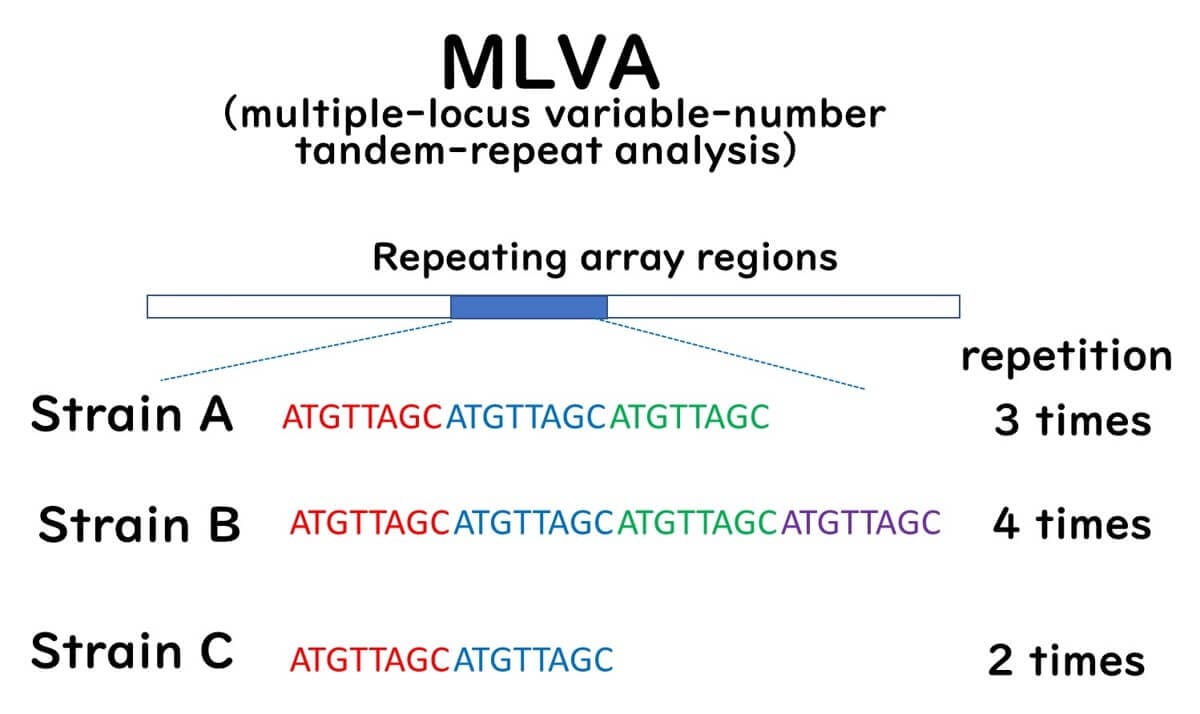
This method employs PCR primers to amplify regions flanking predetermined VNTRs in target bacteria. The PCR products are subsequently analyzed using agarose gel electrophoresis or automated capillary DNA sequencers. The number of tandem repeats is inferred from the size of the PCR products. MLVA profiles, characterized by repeat numbers at VNTR loci, are assigned MLVA type numbers and stored in databases for comparative and epidemiological research.
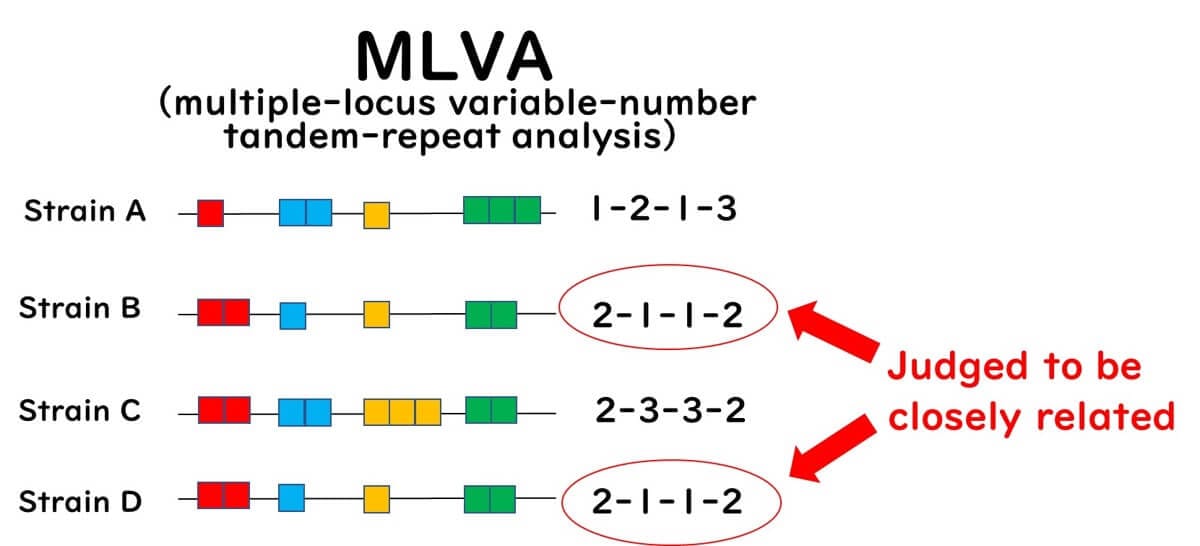
MLVA is a high-resolution technique for detecting genetic differences among closely related strains without relying on next-generation sequencing (NGS), making it a convenient alternative to traditional Sanger-based capillary sequencing. However, since not all bacteria possess repeat sequences, the applicability of MLVA is more limited compared to MLST. Moreover, public health agencies worldwide are progressively adopting NGS-based methods for the molecular epidemiological analysis of foodborne bacteria.
Conclusion
Over the years, bacterial strain typing methods have evolved significantly. While PFGE was once the gold standard, it had limitations in terms of complexity and subjectivity. The introduction of MLST improved data standardization and phylogenetic analysis, making it a powerful tool for global epidemiological studies. Meanwhile, MLVA offered a rapid and high-resolution alternative, particularly suitable for highly clonal bacteria like E. coli O157.
However, both MLST and MLVA have their own limitations, and the field is now shifting toward Whole-Genome Sequencing (WGS) as a comprehensive approach to bacterial typing. As sequencing technology becomes more accessible, WGS will likely complement or replace existing methods, providing even greater accuracy in tracking and understanding bacterial evolution.
Related posts:
Real-Time PCR in Food Safety: Practical Insights into the TaqMan Method Real-Time PCR, particularly the TaqMan method, is transforming microbiological testing...
Real-Time PCR in Food Microbiology: Challenges and Applications Real-Time PCR has revolutionized diagnostic testing in various fields, including...
PCR Testing in Food Microbiology: Key Principles, Benefits, and Applications PCR testing in food microbiology offers unmatched advantages over traditional...
Mastering PCR Primer Design for Accurate Microbial Detection in Food Microbiology PCR primer design is a cornerstone of accurate and reliable...

Discover expert-led lessons in food microbiology designed for professionals and beginners alike.

Author of this Blog: Bon Kimura
Bon Kimura, Professor Emeritus at Tokyo University of Marine Science and Technology (TUMSAT), specializes in food microbiology. He obtained his PhD from Kyoto University and became a professor at TUMSAT in 2006, serving as Dean of the Faculty of Marine Science from 2012-2015. Kimura has published over 200 international papers on food safety, pathogens, and spoilage bacteria. He has received multiple awards, including the Japanese Society for Food Microbiology Award (2019). Kimura also served as an editor for the International Journal of Food Microbiology from 2012 to 2024, where he was the principal reviewer for 1,927 papers.
Copyright © Introduction to Food Microbiology and Safety All Rights Reserved.