Introduction to Food Microbiology and Safety
What is PCR? Basics, Process, and Applications in Food Microbiology Testing
PCR (Polymerase Chain Reaction) is a revolutionary technique for amplifying DNA, making it an essential tool in food microbiology testing. This article provides a clear explanation of PCR, its process, and its critical role in ensuring food safety.
What is PCR (Polymerase Chain Reaction)?
PCR, or Polymerase Chain Reaction, is a groundbreaking technique used to amplify specific DNA sequences of an organism within a small plastic tube. This method uses short DNA fragments called primers, which flank the target DNA region, along with a template-specific synthesis reaction catalyzed by DNA polymerase. Even with an extremely small amount of template DNA, PCR can amplify the target region by 100,000 to 1,000,000 times in a matter of hours.
This powerful amplification ability makes PCR indispensable for applications such as detecting microorganisms in food samples, ensuring food safety, and conducting microbiological testing with high precision.
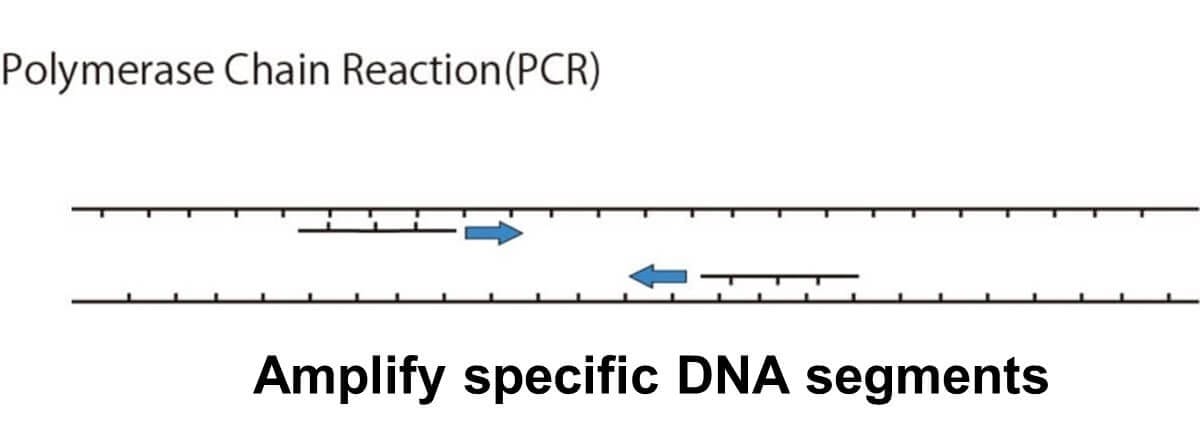
The PCR Process
The PCR process consists of a series of steps designed to exponentially amplify a target DNA sequence. Below is a simplified explanation:
Preparation:
A PCR tube (a small plastic tube with a typical capacity of 0.6 ml) is prepared by loading the following components:
Sample DNA extracted from food,
Two primers complementary to the target DNA sequence,
Taq DNA polymerase (an enzyme responsible for DNA amplification),
A mixture of nucleotides (the building blocks for DNA synthesis).
These components are combined using a micropipette.
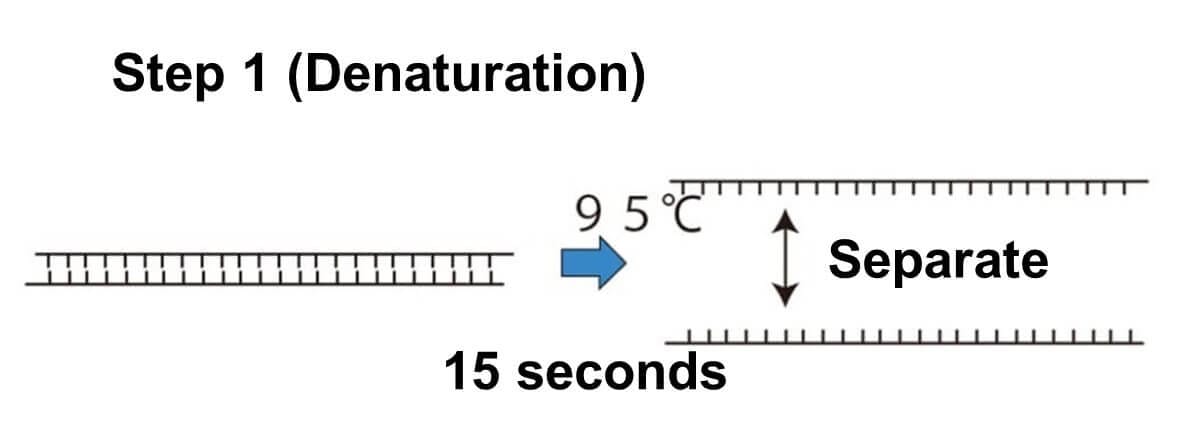
Denaturation:
The PCR tube is heated to 90–95°C for approximately 60 seconds. This high temperature causes the double-stranded DNA to separate into single strands by breaking the weak hydrogen bonds between the base pairs.
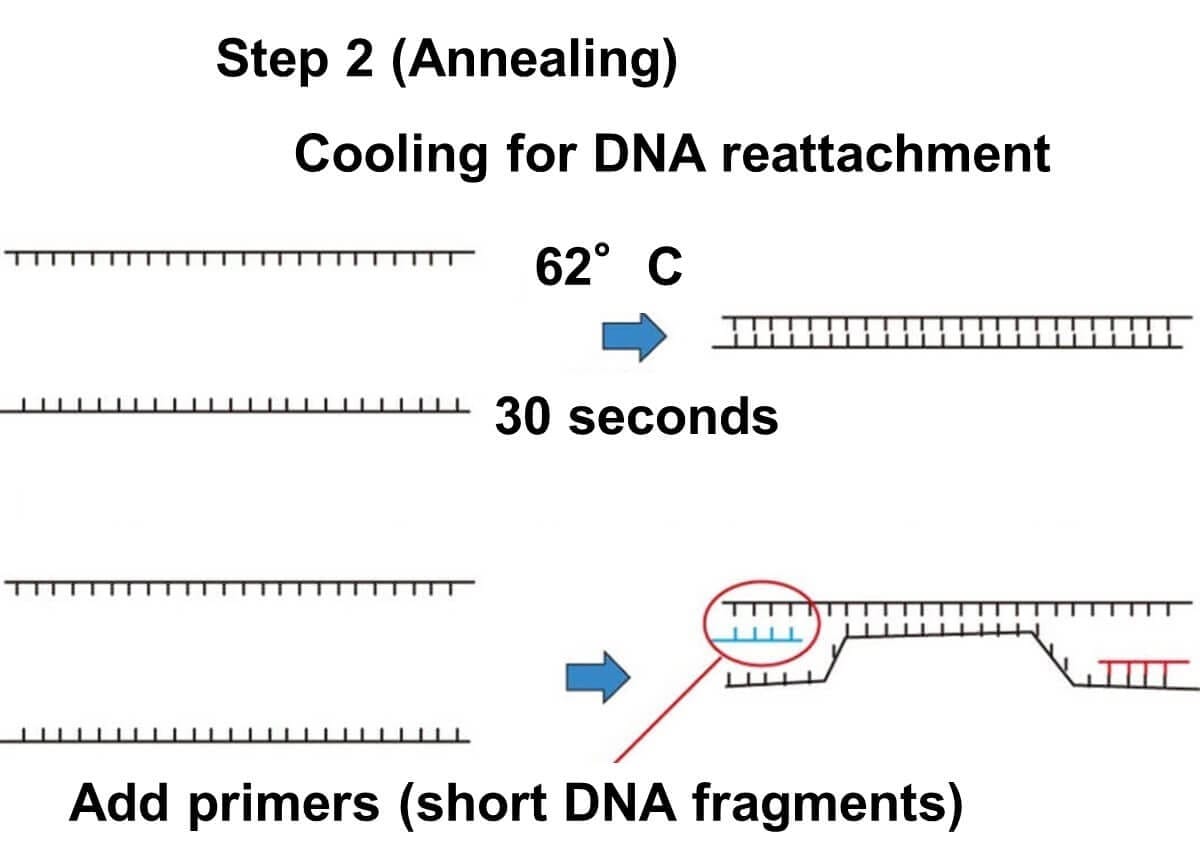
Annealing:
The temperature is then lowered to 40–60°C for about 60 seconds. During this stage, the primers bind to their complementary sequences on the single-stranded DNA target regions. This step is called "annealing," a term borrowed from metallurgy, where it refers to heating and cooling materials to alter their properties. In the PCR process, it specifically refers to the reformation of double-stranded DNA with the primers attached.
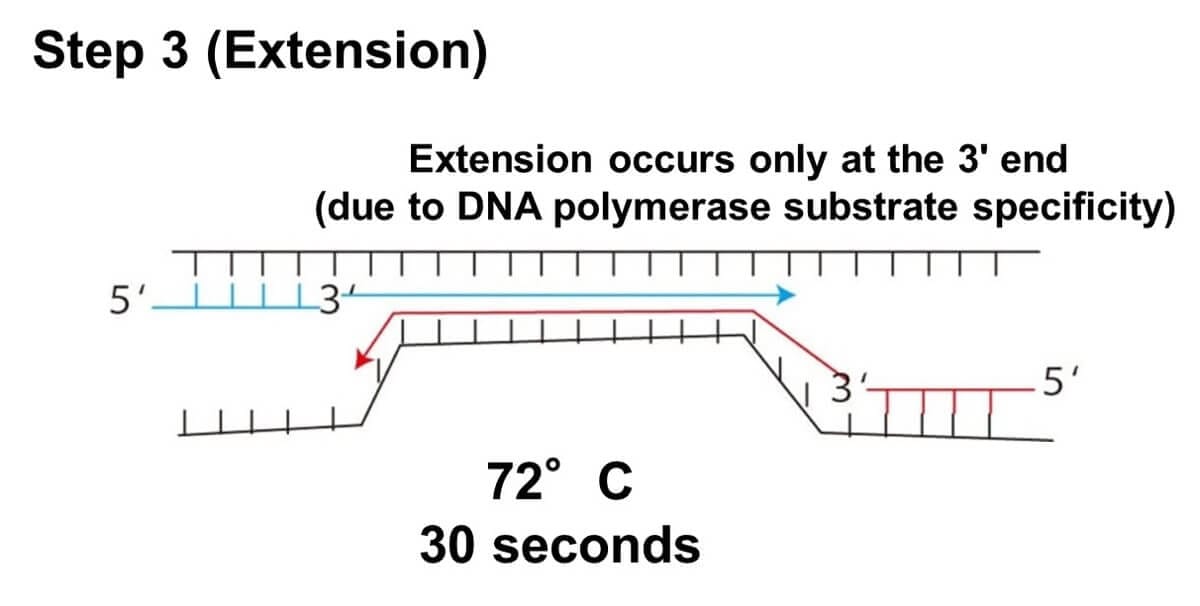
Extension:
At 70–75°C, Taq DNA polymerase synthesizes a new DNA strand complementary to the template DNA strand. This step takes approximately 60 seconds.
Cycling:
The above three steps (denaturation, annealing, and extension) are repeated multiple times, typically 20–40 cycles, using a thermal cycler. Each cycle exponentially amplifies the target DNA fragment, resulting in millions of copies within a few hours.

PCR vs. Bacterial Cell Division
Bacterial cell division is a complex process where the entire genome, consisting of approximately 10^6 bases, is replicated. In addition to DNA replication, the cell must synthesize proteins, lipids, and carbohydrates essential for forming a new cell. Even under optimal conditions, the fastest-growing bacteria require 20–30 minutes to complete one division cycle.
In contrast, PCR is highly efficient. Each cycle, lasting about 3 minutes, doubles the target DNA sequence (usually a few hundred bases in length). This exponential amplification allows PCR to generate millions of copies of the target gene within 2–3 hours. By comparison, traditional bacterial culture methods may take at least 24 hours to grow sufficient cells for detection.
The rapidity of PCR makes it a transformative tool in food microbiology, enabling the timely detection of foodborne pathogens and ensuring food safety more efficiently than conventional methods.
Understanding DNA Properties for PCR Principles
To fully understand PCR, it is important to grasp the fundamental properties of DNA. The following sections explain the DNA characteristics that make PCR possible.
Why Cycle Between 95°C and 62°C?
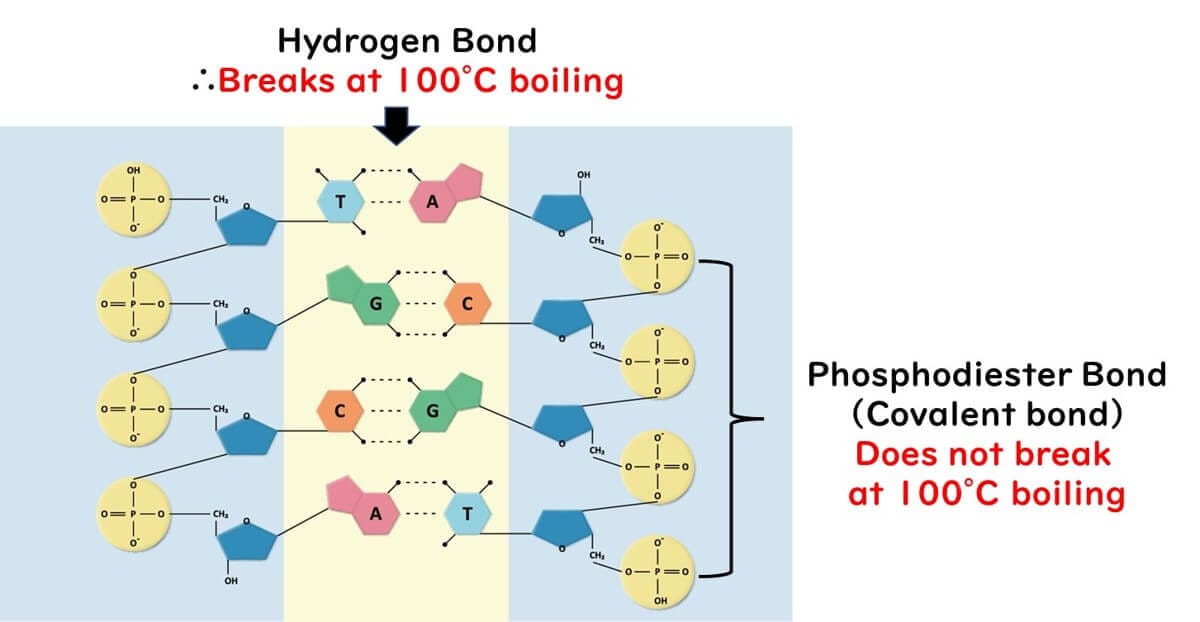
DNA has a double-helix structure, where two chains of deoxyribose molecules are linked by phosphate bonds. These chains are held together by hydrogen bonds between complementary bases: adenine (A) pairs with thymine (T), and cytosine (C) pairs with guanine (G). This arrangement forms the rungs of a ladder-like structure.
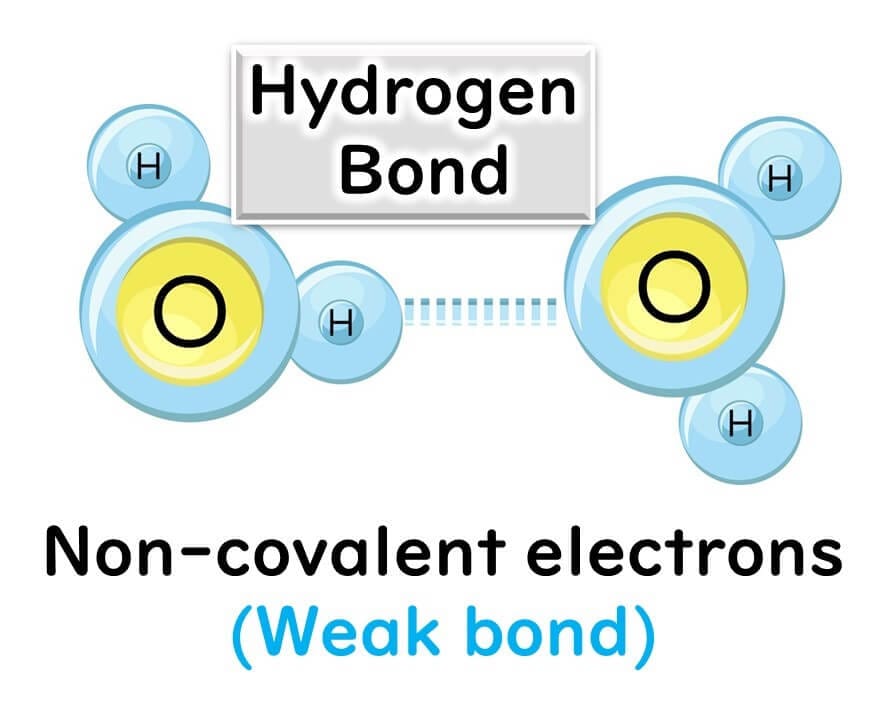
Hydrogen Bonds:
The hydrogen bonds holding the bases together are relatively weak, making it possible to break them by heating to 90–95°C during the denaturation step of PCR. This separation results in single-stranded DNA.
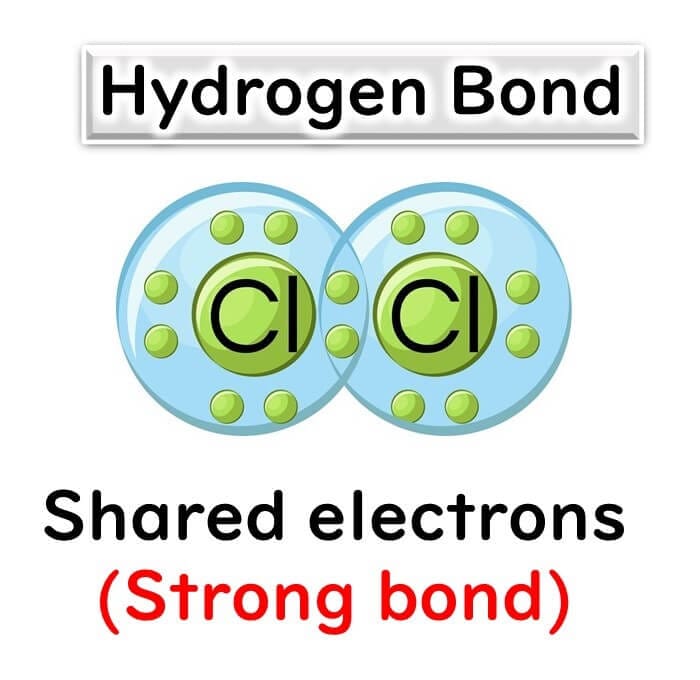
Covalent Bonds:
The phosphate bonds linking the deoxyribose molecules in the DNA backbone are covalent and much stronger. These bonds remain intact even at high temperatures, ensuring the stability of the DNA strands during PCR.
This combination of weak hydrogen bonds and strong covalent bonds allows for the precise temperature control required in PCR, enabling strand separation without compromising the DNA's structural integrity.
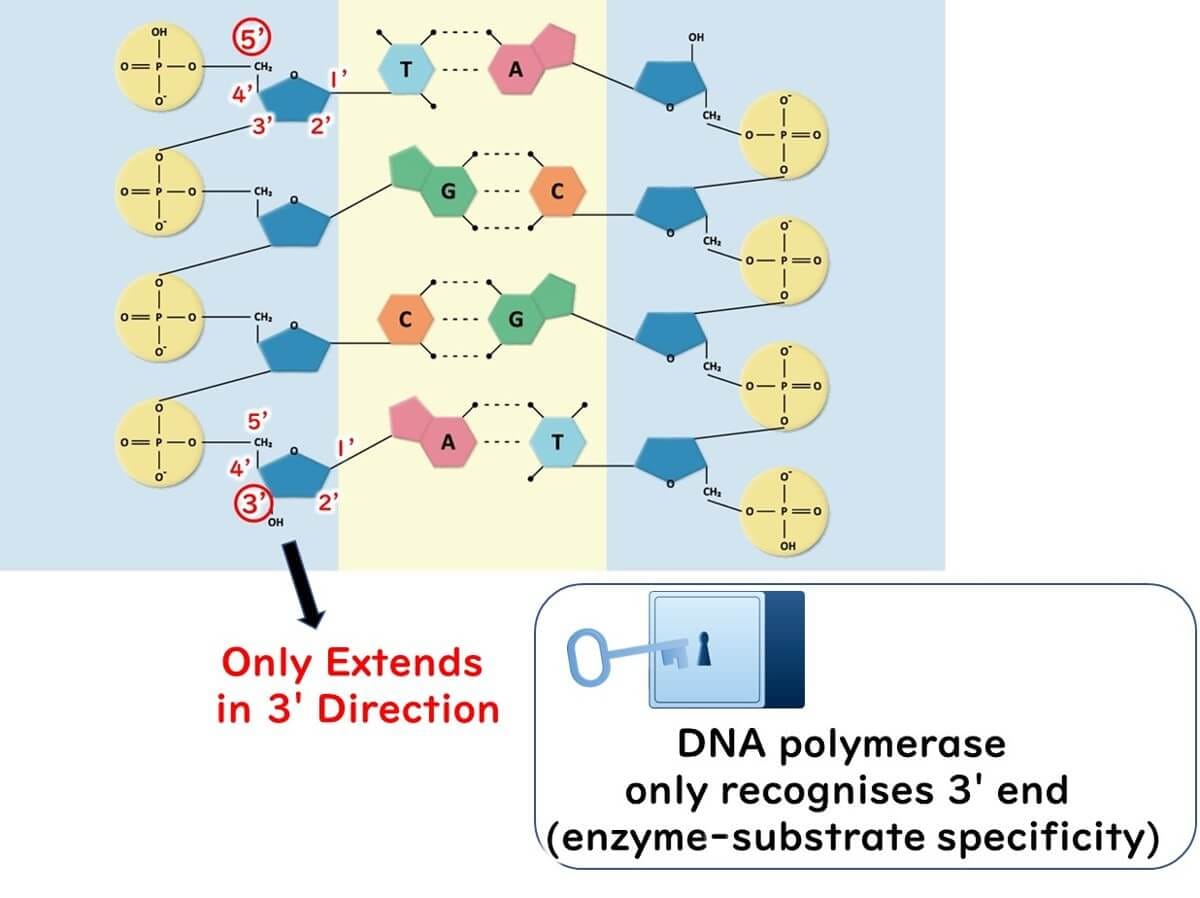
Why Does DNA Extension Occur Only at the 3' End?
The directionality of DNA synthesis is a crucial factor in PCR. DNA polymerase can only extend a DNA strand from the 3' end. Here’s why:
Specificity of DNA Polymerase:
Enzymes like DNA polymerase function based on a lock-and-key mechanism. The enzyme recognizes the 3' end of the deoxyribose molecule in DNA but cannot interact with the 5' end due to its molecular structure.
Deoxyribose Structure:
The deoxyribose in DNA has carbon atoms numbered 1' to 5'. DNA polymerase attaches nucleotides to the hydroxyl group (-OH) present at the 3' carbon. The 5' end lacks this functional group, preventing DNA polymerase from extending in that direction.
This directional limitation ensures the controlled synthesis of new DNA strands during the PCR process.
Why Does DNA Polymerase Work at High Temperatures Like 72°C?
Most enzymes denature and lose activity at temperatures above 65°C. DNA polymerase, however, must remain active at higher temperatures to function during PCR. This requirement led to the discovery of Taq polymerase, an enzyme that is stable and effective at high temperatures.
Heat Stability:
Taq polymerase, derived from the thermophilic bacterium Thermus aquaticus, remains active across a wide temperature range, including the 70–75°C required for the extension step of PCR. This characteristic is essential for PCR's efficiency and reliability.
For more details on protein denaturation and heat stability, see the related article on
The Discovery of Taq Polymerase
Before the advent of modern PCR, DNA polymerase from Escherichia coli (E. coli) was commonly used. However, this enzyme became inactive at temperatures above 65°C, necessitating the addition of fresh enzyme for every PCR cycle—a cumbersome and inefficient process.
Breakthrough with Taq Polymerase:
Taq polymerase was isolated from Thermus aquaticus, a thermophilic bacterium found in the hot springs of Yellowstone National Park, USA. Its remarkable heat stability allows it to function across the temperature range of 65–100°C, making it ideal for PCR. This discovery revolutionized the field, enabling the development of the efficient PCR technology used today.

Detecting PCR Amplification (Electrophoresis)
Before the development of real-time PCR approximately 20 years ago, electrophoresis was the primary method for detecting whether target DNA had been successfully amplified. Despite advances in technology, electrophoresis remains widely used in laboratories worldwide due to its simplicity and reliability.
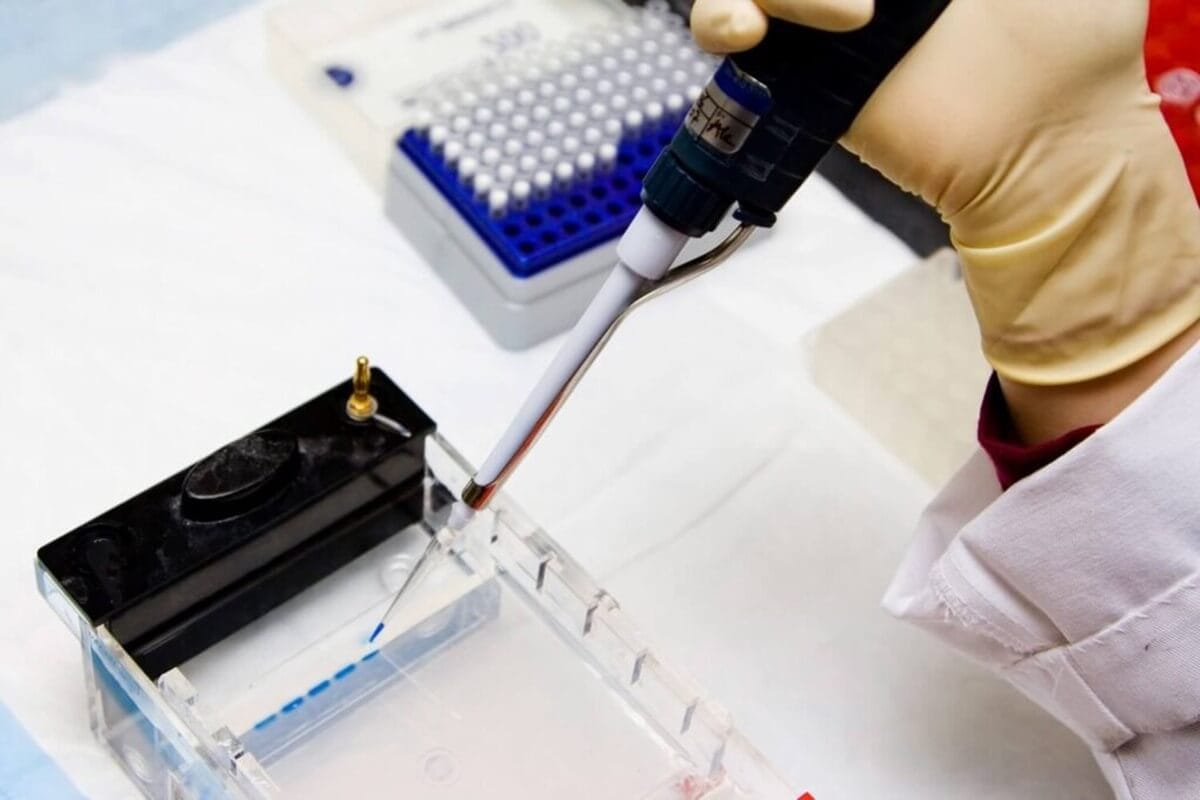
The Electrophoresis Process
Loading the Gel:
Amplified DNA from the PCR reaction is loaded into wells within an agarose gel. DNA molecules, composed of phosphate bonds, carry a negative charge due to the phosphorus component. This negative charge allows DNA to move under an electric current.
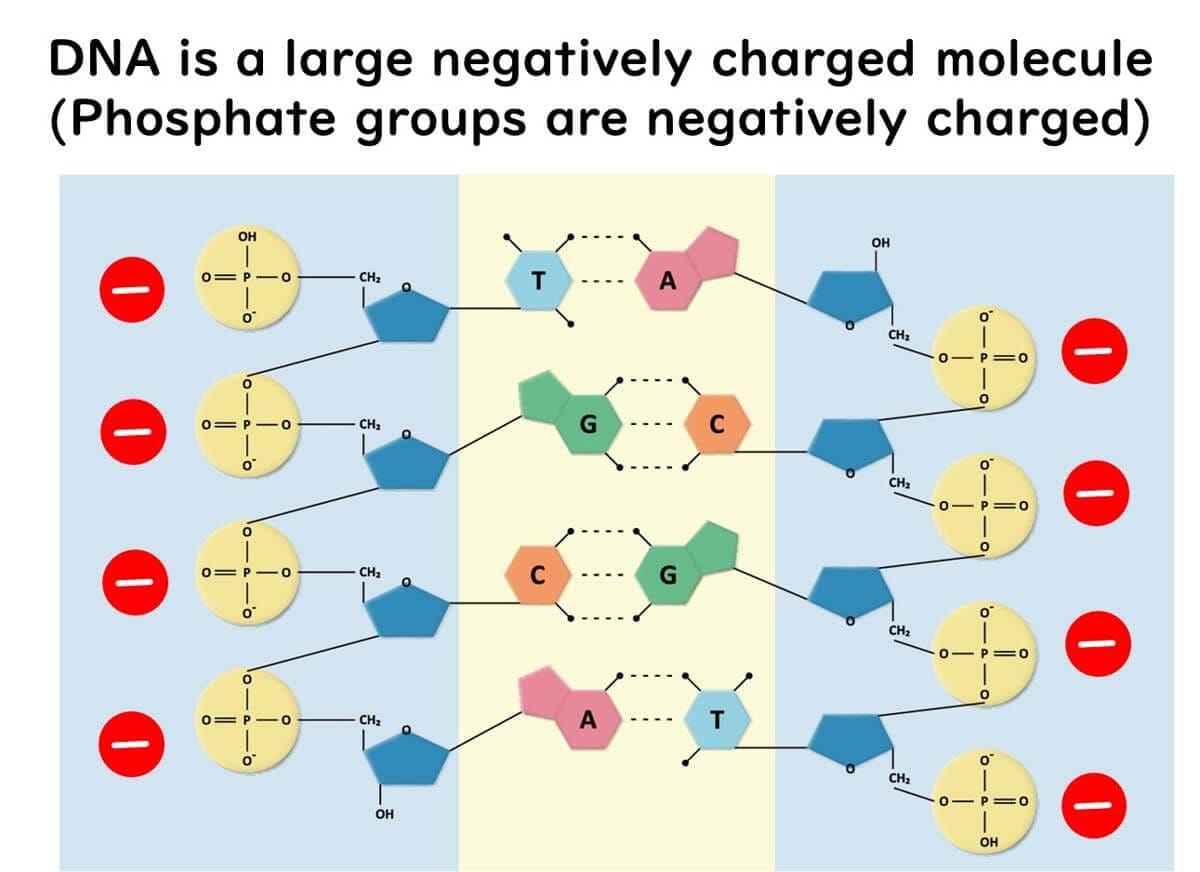
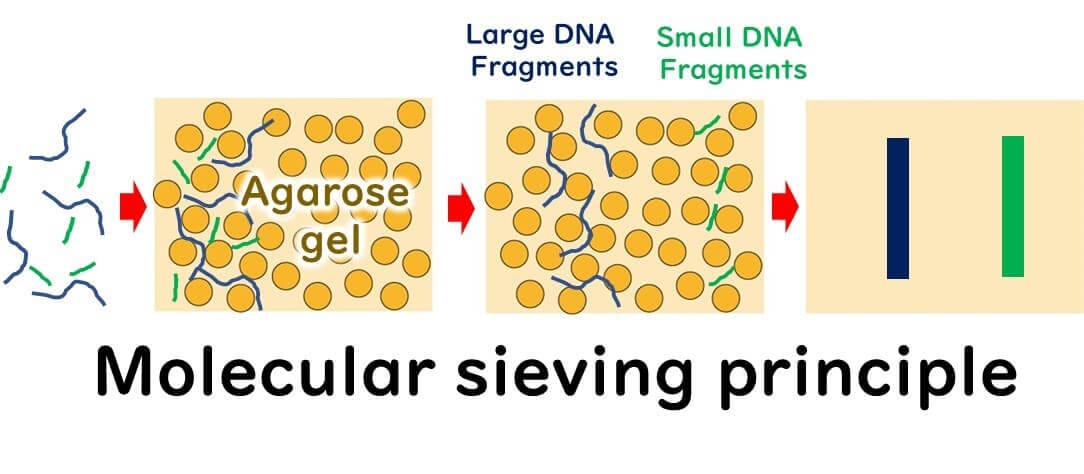
2. Running the Gel:
When an electric current is applied, the negatively charged DNA molecules migrate toward the positive electrode. Smaller DNA fragments pass through the gel matrix more easily and travel faster than larger fragments, resulting in size-based separation.
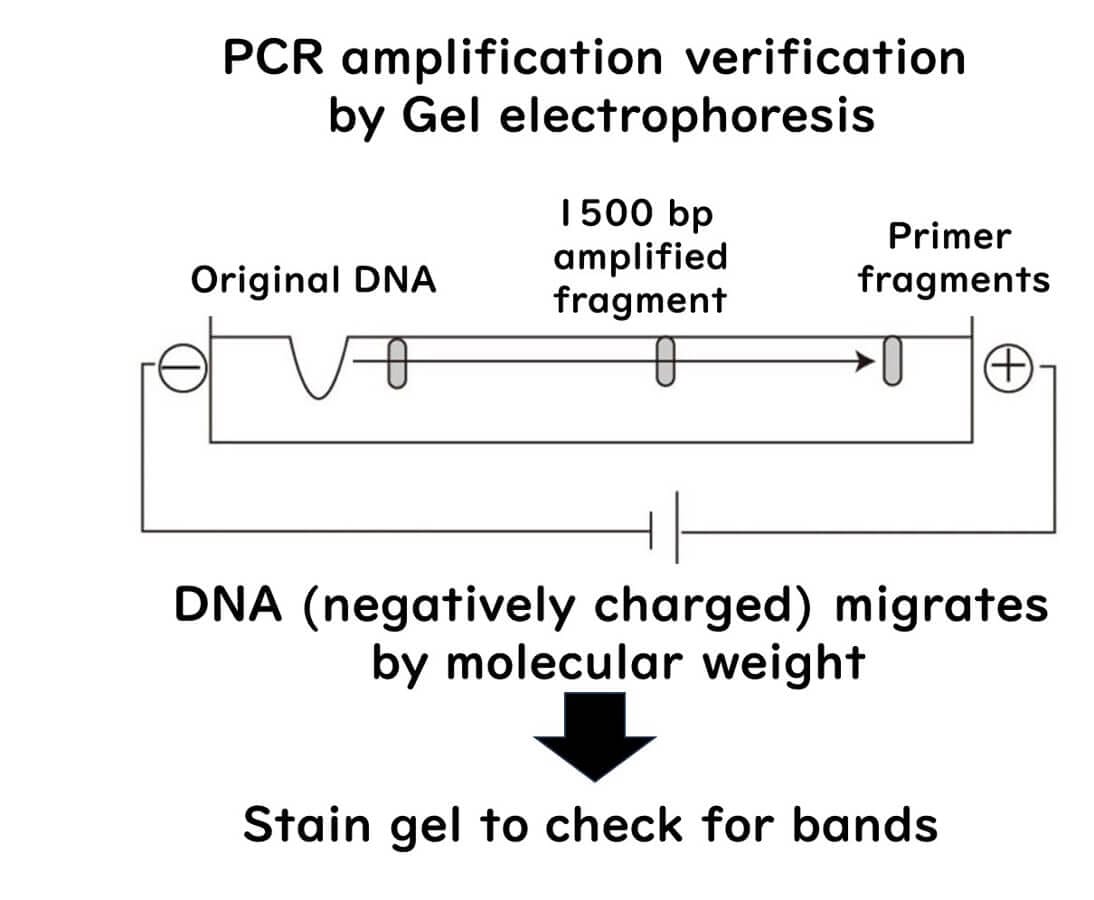
3. Analyzing the Results:
After a set amount of time, the DNA fragments are visualized within the gel at different distances corresponding to their sizes. For instance:
If the target DNA fragment is 1500 base pairs (bp) in size and is detected at the position corresponding to 1500 bp, the PCR amplification was successful.
Conversely, if no DNA is detected at this position, the PCR reaction is considered negative.
This straightforward approach allows researchers to confirm the presence or absence of the target DNA and evaluate the efficiency of the PCR process.

Discover expert-led lessons in food microbiology designed for professionals and beginners alike.

Author of this Blog: Bon Kimura
Bon Kimura, Professor Emeritus at Tokyo University of Marine Science and Technology (TUMSAT), specializes in food microbiology. He obtained his PhD from Kyoto University and became a professor at TUMSAT in 2006, serving as Dean of the Faculty of Marine Science from 2012-2015. Kimura has published over 200 international papers on food safety, pathogens, and spoilage bacteria. He has received multiple awards, including the Japanese Society for Food Microbiology Award (2019). Kimura also served as an editor for the International Journal of Food Microbiology from 2012 to 2024, where he was the principal reviewer for 1,927 papers.
Copyright © Introduction to Food Microbiology and Safety All Rights Reserved.